Safe before sale
Learnings from the FDA’s model of life sciences oversight for foundation models
14 December 2023
Reading time: 150 minutes

Executive summary
What can foundation model oversight learn from the US Food and Drug Administration (FDA)?
In the last year, policymakers around the world have grappled with the challenge of how to regulate and govern foundation models – artificial intelligence (AI) models like OpenAI’s GPT-4 that are capable of a range of general tasks such as text synthesis, image manipulation and audio generation. Policymakers, civil society organisations and industry practitioners have expressed concerns about the reliability of foundation models, the risk of misuse of their powerful capabilities and the systemic risks they could pose as more and more people begin to use them in their daily lives.
Many of these risks to people and society – such as the potential for powerful and widely used AI systems to discriminate against particular demographics, or to spread misinformation more widely and easily – are not new, but foundation models have some novel features that could greatly amplify the potential harms.
These features include their generality and ability to complete range of tasks; the fact that they are ‘built on’ for a wide range of downstream applications, creating a risk that a single point of failure could lead to networked catastrophic consequences; fast and (sometimes) unpredictable jumps in their capabilities and behaviour, which make it harder to foresee harm; and their wide-scale accessibility, which puts powerful AI capabilities in the hands of a much larger number of people.
Both the UK and US governments have released voluntary commitments for developers of these models, and the EU’s AI Act includes some stricter requirements for models before they can be sold on the market. The US Executive Order on AI also includes some obligations on some developers of foundation models to test their systems for certain risks.[1] [2]
Experts agree that foundation models need additional regulatory oversight due to their novelty, complexity and lack of clear safety standards. Oversight needs to enable learning about risks, and to ensure iterative updates to safety assessments and standards.
Notwithstanding the unique features of foundation models, this is not the first time that regulators have grappled with how to regulate complex, novel technologies that raise a variety of sociotechnical risks.[3] One area where this challenge already exists is in life sciences. Drug and medical device regulators have a long history of applying a rigorous oversight process to novel, groundbreaking and experimental technologies that – alongside their possible benefits – could present potentially severe consequences for people and society.
This paper draws on interviews with 20 experts and a literature review to examine the suitability and applicability of the US Food and Drug Administration (FDA) oversight model to foundation models. It explores the similarities and differences between medical devices and foundation models, the limitations of the FDA model as applied to medical devices, and how the FDA’s governance framework could be applied to the governance of foundation models.
This paper highlights that foundation models may pose risks to the public that are similar to — or even greater than — Class III medical devices (the FDA’s highest risk category). To begin to address the mitigation of these risks through the lens of the FDA model, the paper lays out general principles to strengthen oversight and evaluation of the most capable foundation models, along with specific recommendations for each layer in the supply chain.
This report does not address questions of international governance implications, the political economy of the FDA or regulating AI in medicine specifically. Rather, this paper seeks to answer a simple question: when designing the regulation of complex AI systems, what lessons and approaches can regulators draw on from medical device regulation?
A note on terminology
Regulation refers to the legally binding rules that govern the industry, setting the standards, requirements and guidelines that must be complied with.
Oversight refers to the processes of monitoring and enforcing compliance with regulations, for example through audits, reporting requirements or investigations.
What is FDA oversight?
With more than one hundred years’ history, a culture of continuous learning, and increasing authority, the FDA is a long-established regulator, with FDA-regulated products accounting for about 20 cents of every dollar spent by US consumers.
The FDA regulates drugs and medical devices by assigning them a specific risk level corresponding to how extensive subsequent evaluations, inspections and monitoring will be at different stages of development and deployment. The more risky and more novel a product, the more tests, evaluation processes and monitoring it will undergo.
The FDA does this by providing guidance and setting requirements for drug and device developers to follow, including regulatory approval of any protocols the developer will use for testing, and evaluating the safety and efficacy of the product.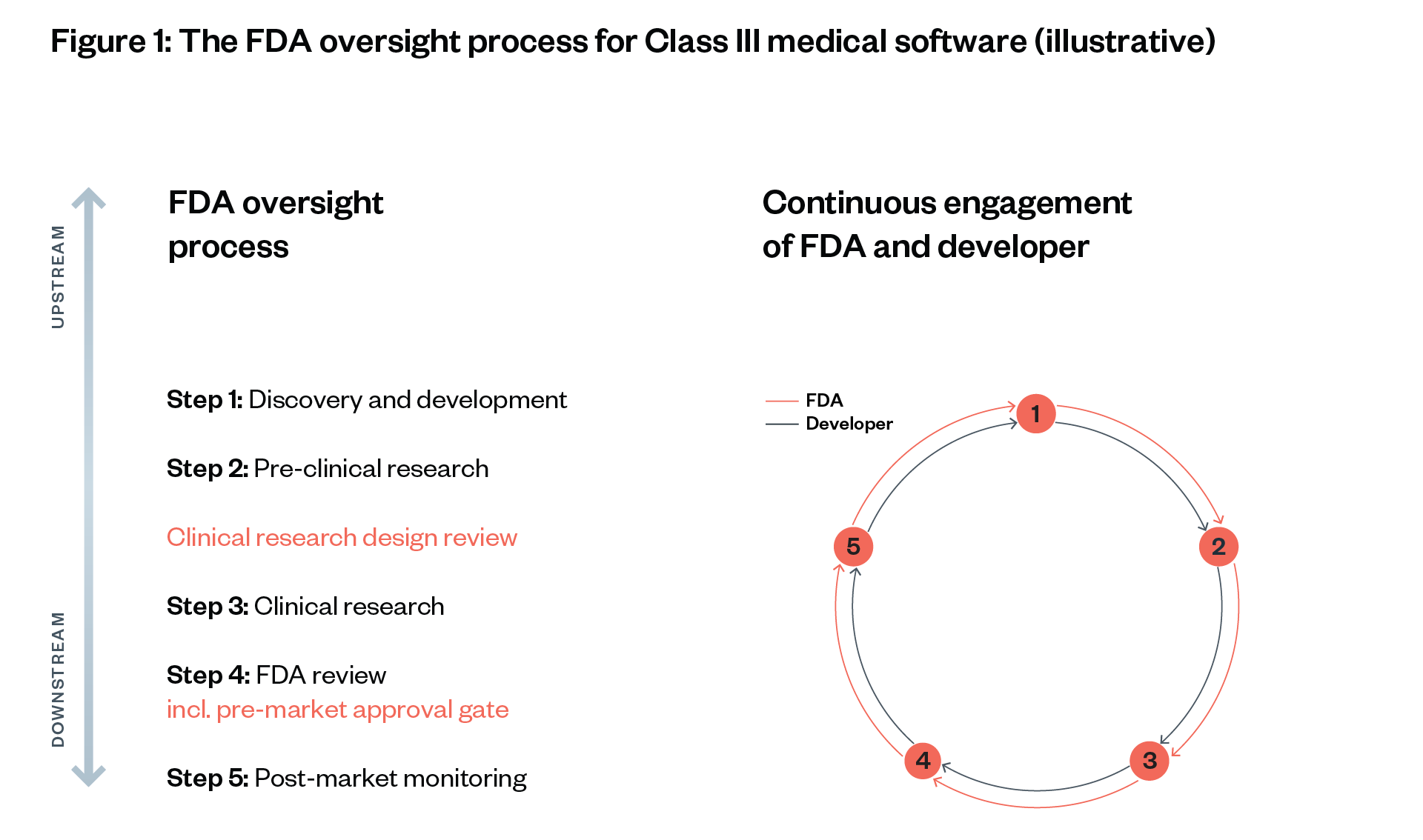
The FDA has extensive auditing powers, with the ability to inspect drug companies’ data, processes and systems at will. It also requires companies to report incidents, failures and adverse impacts to a central registry. There are substantial fines for failing to follow appropriate regulatory guidance, and the FDA has a history of enforcing these sanctions.
Core risk-reducing aspects of FDA oversight
- Risk- and novelty-driven oversight: The riskier and more novel a product, the more tests, evaluation processes and monitoring there will be.
- Continuous, direct engagement with developers from development through to market: Developers must undergo a rigorous testing process through a protocol agreed with the FDA.
- Wide-ranging information access: The FDA has statutory powers to access comprehensive information, for example, clinical trial results and patient data.
- Burden of proof on developers: Developers must demonstrate the efficacy and safety of a drug or medical device at various ‘approval gates’ before the product can be tested on humans or be sold on a market.
- Balancing innovation with efficacy and safety: This builds acceptance for the FDA’s regulatory authority.
How suitable is FDA-style oversight for foundation models?
Our findings show that foundation models are at least as complex as and more novel than FDA Class III medical devices (the highest risk category), and that the risks they pose are potentially just as severe.[4][5][6] Indeed, the fact that these models are deployed across the whole economy, interacting with millions of people, means that they are likely to pose systemic risks far beyond those of Class III medical devices.[7] However, the exact risks of these models are so far not fully clear. Risk mitigation measures are uncertain and risk modelling is poor or non-existent.
The regulation of Class III medical devices offers policymakers valuable insight into how they might regulate foundation models, but it is also important that they are aware of the limitations.
Limitations of FDA-style oversight for foundation models
- High cost of compliance: A high cost of compliance could limit the number of developers, which may benefit existing large companies. Policymakers may need to consider less restrictive requirements for smaller companies that have fewer users, coupled with support for such companies in compliance and via streamlined regulatory pathways.
- Limited range of risks assessed: The FDA model may not be able to fully address the systemic risks and the risks of unexpected capabilities associated with foundation models. Medical devices are not general purpose, and the FDA model therefore largely assesses efficacy and safety in narrow contexts. Policymakers may need to create new, exploratory methods for assessing some types of risk throughout the foundation model supply chain, which may require increased post-market monitoring obligations.
- Overreliance on industry: Regulatory agencies like the FDA sometimes need industry expertise, especially in novel areas where clear benchmarks have not yet been developed and knowledge is concentrated in industry. Foundation models present a similar challenge. This could raise concerns around regulatory capture and conflicts of interest. An ecosystem of independent academic and governmental experts needs to be built up to support balanced, well-informed oversight of foundation models, with clear mechanisms for those impacted by AI technologies to contribute. This could be at the design and development stage, eliciting feedback from pre-market ‘sandboxing’, or through market approval processes (under the FDA regime, patient representatives have a say in this process). At any step in the process, consideration should be given to who is involved (this could range from a representative panel to a jury of members of the public), the depth of engagement (from public consultations through to partnership decision-making), and methods (for example, from consultative exercises such as focus groups, to panels and juries for deeper engagement).
General principles for AI regulators
To strengthen oversight and evaluations of the most capable foundation models (for example, OpenAI’s GPT-4), which currently lag behind FDA oversight in aspects of risk-reducing external scrutiny:
- Establish continuous, risk-based evaluations and audits throughout the foundation model supply chain.
- Empower regulatory agencies to evaluate critical safety evidence directly, supported by a third-party ecosystem – consistently proven higher quality than self- or second-party evaluations across industries.
- Ensure independence of regulators and external evaluators, through mandatory industry fees and a sufficient budget for regulators that contract third parties. While existing sector-specific regulators, for example, the Consumer Financial Protection Bureau (CFPB) in the USA, may review downstream AI applications, there might be a need for an upstream regulator of foundation models themselves. The level of funding for such a regulator would need to be similar to that of other safety-critical domains, such as medicine.
- Enable structured access to foundation models and adjacent components for evaluators and civil society. This will help ensure the technology is designed and deployed in a manner that meets the needs of the people who are impacted by its use, and enable methods to offer accountability mechanisms if it is not
- Enforce a foundation model pre-approval process, shifting the burden of proof to developers.
Recommendations for AI regulators, developers and deployers

Data and compute layers oversight
- Regulators should compel pre-notification of, and information-sharing on, large training runs.
- Regulators should compel mandatory model and dataset documentation and disclosure for the pre-training and fine-tuning of foundation models,[8] [9] [10] including a capabilities evaluation and risk assessment within the model card for the (pre-) training stage and post-market.
Foundation model layer oversight
- Regulators should introduce a pre-market approval gate for foundation models, as this is the most obvious point at which risks can proliferate. In any jurisdiction, defining the approval gate will require significant work, with input from all relevant stakeholders. In critical or high-risk areas, depending on the jurisdiction and existing or foreseen pre-market approval for high-risk use, regulators should introduce an additional approval gate at the application layer of the supply chain.
- Third-party audits should be required as part of the pre-market approval process, and sandbox testing in real-world conditions should be considered.
- Developers should enable detection mechanisms for the outputs of generative foundation models.
- As part of the initial risk assessment, developers and deployers should document and share planned and foreseeable modifications throughout the foundation model’s supply chain.
- Foundation model developers, and high-risk application providers building on top of these models, should enable an easy complaint mechanism for users to swiftly report any serious risks that have been identified.
Application layer oversight
- Existing sector-specific agencies should review and approve the use of foundation models for a set of use cases, by risk level.
- Downstream application providers should make clear to end users and affected persons what the underlying foundation model is, including if it is an open-source model, and provide easily accessible explanations of systems’ main parameters and any opt-out mechanisms or human alternatives available.
Post-market monitoring
- An AI ombudsman should be considered, to take and document complaints or known instances of harms of AI. This should be complimented by a comprehensive remedies framework for affected persons based on clear avenues for redress.
- Developers and downstream deployers should provide documentation and disclosure of incidents throughout the supply chain, including near misses. This could be strengthened by requiring downstream developers (building on top of foundation models at the application layer) and end users (for example, medical or education professionals) to also disclose incidents.
- Foundation model developers, downstream deployers and hosting providers (for example GitHub or Hugging Face) should be compelled to restrict, suspend or retire a model from active use if harmful impacts, misuse or security vulnerabilities (including leaks or otherwise unauthorised access) arise.
- Host layer actors (for example cloud service providers or model hosting platforms) should also play a role in evaluating model usage and implementing trust and safety policies to remove harmful models that have demonstrated or are likely to demonstrate serious risks, and flagging harmful models to regulators when it is not in their power to take them down.
- AI regulators should have strong powers to investigate and require evidence generation from foundation model developers and downstream deployers. This should be strengthened by whistleblower protections for any actor involved in development or deployment who raises concerns about risks to health or safety.
- Any regulator should be funded to a level comparable to (if not greater than) regulators in other domains where safety and public trust are paramount and where underlying technologies form part of national infrastructure – such as civil nuclear, civil aviation, medicines, or road and rail.[11] Given the level of resourcing required, this may be partly funded by AI developers over a certain threshold.
- The law around AI liability should be clarified to ensure that legal and financial liability for AI risk is distributed proportionately along foundation model supply chains.
Introduction
As governments around the world consider the regulation of artificial intelligence (AI), many experts are suggesting that lessons should be drawn from other technology areas. The US Food and Drug Administration (FDA) and its approval process for drug development and medical devices is one of the most cited areas in this regard.
This paper seeks to understand if and how FDA-style oversight could be applied to AI, and specifically to foundation models, given their complexity, novelty and potentially severe risk profile – each of which arguably exceeds those of the products regulated by the FDA.
This paper first maps the FDA review process for Class III medical software, to identify both the risk-reducing features and the limitations of FDA-style oversight. It then considers the suitability and applicability of FDA processes to foundation models and suggests how FDA risk-reducing features could be applied across the foundation model supply chain. It concludes with actionable recommendations for policymakers.
What are foundation models?
Foundation models are a form of AI system capable of a range of general tasks, such as text synthesis, image manipulation and audio generation.[12] Notable examples include OpenAI’s GPT-4 – which has been used to create products such as ChatGPT – and Anthropic’s Claude 2.
Advances in foundation models raise concerns about reliability, misuse, systemic risks and serious harms. Developers and researchers of foundation models have highlighted that their wide range of capabilities and unpredictable behaviours[13] could pose a series of risks, including:
- Accidental harms: Foundation models can generate confident but factually incorrect statements, which could exacerbate problems of misinformation. In some cases this could have potentially fatal consequences, for example, if someone is misled into eating something poisonous or taking the wrong medication.[14] [15]
- Misuse harms: These models could enable actors to intentionally cause harm, from harassment[16] through to cybercrime at a greater scale[17] or biosecurity risks.[18] [19]
- Structural or systemic harms: If downstream developers increasingly rely on foundation models, this creates a single point of dependency on a model, raising security risks.[20] It also concentrates market power over cutting-edge foundation models as few private companies are able to develop foundation models with hundreds of millions of users.[21] [22] [23]
- Supply chain harms: These are harms involving the processes and inputs used to develop AI, such as poor labour practices, environmental impacts and the inappropriate use of personal data or protected intellectual property.[24]
Context and environment
Experts agree that foundation models are a novel technology in need of additional oversight. This sentiment was shared by industry, civil society and government experts at an Ada Lovelace Institute roundtable on standards-setting held in May 2023. Attendees largely agreed that foundation models represent a ‘novel’ technology without an established ‘state of the art’ for safe development and deployment.
This means that additional oversight mechanisms may be needed, such as testing the models in a ‘sandbox’ environment or regular audits and evaluations of a model’s performance before and after its release (similar to the approach to the testing, approval and monitoring approaches in public health). Such mechanisms would enable greater transparency and accessibility for actors with incentives more aligned with societal interest in assessing (second order) effects on people.[25]
Crafting AI regulation is a priority for governments worldwide. In the last three years, national governments across the world have sought to draft legislation to regulate the development and deployment of AI in different sectors of society.
The European AI Act takes a risk-based approach to regulation, with stricter requirements applying to AI models and systems that pose a high risk to health, safety or fundamental rights. In contrast, the UK has proposed a principles-based approach, calling for existing individual regulators to regulate AI models through an overarching set of principles.
Policymakers in the USA have proposed a different approach in the Algorithm Accountability Act,[26] which would create a baseline requirement for companies building foundation models and AI systems to assess the impacts of ‘automating critical decision-making’ and empower an existing regulator to enforce this requirement. Neither the UK nor the USA have ruled out ‘harder’ regulation that would require the creation of a new (or empowering an existing) body for enforcement.
Regulation in public health, such as FDA pre-approvals, can inspire AI regulation. As governments seek to develop their approach to regulating AI, they have naturally turned to other emerging technology areas for guidance. One area routinely mentioned is the regulation of public health – specifically, the drug development and medical device regulatory approval process used by the FDA.
The FDA’s core objective is to ‘speed innovations that make food and drug products more effective, safer and more affordable’ to ‘maintain and improve the public’s health’. In practice, the FDA model requires developers of drugs or medical devices to provide (sufficiently positive) evidence on the safety risks, efficacy and accessibility of products before they are approved to be sold in a market or continue to the next development phase (referred to as pre-market approval or pre-approval).
Many call for FDA-style oversight for AI, though its detailed applicability for foundation models is largely unexamined. Applying lessons from the FDA to AI is not a new idea,[27] [28] [29] though it has recently gained significant traction. In a May 2023 Senate Hearing, renowned AI expert Gary Marcus testified that priority number one should be ‘a safety review like we use with the FDA prior to widespread deployment’.[30] Leading AI researchers Stuart Russell and Yoshua Bengio have also called for FDA-style oversight of new AI models.[31] [32] [33] In a recent request for evidence by the USA’s National Telecommunications and Information Administration on AI accountability mechanisms, 43 pieces of evidence mentioned the FDA as an inspiration for AI oversight.[34]
However, such calls often lack detail on how appropriate the FDA model is to regulate AI. The regulation of AI for medical purposes has received extensive attention,[35] [36] but there has not yet been a detailed analysis on how FDA-style oversight could be applied to foundation models or other ‘general-purpose’ AI.
Drug regulators have a long history of applying a rigorous oversight process to novel, groundbreaking and experimental technologies that – alongside their possible benefits – present potentially severe consequences.
Such technologies include gene editing, biotechnology and medical software. As with drugs, the effects of most advanced AI models are largely unknown but potentially significant.[37] Both public health and AI are characterised by fast-paced research and development progress, the complex nature of many components, their potential risk to human safety, and the uncertainty of risks posed to different groups of people.
As market sectors, public health and AI are both dominated by large private-sector organisations developing and creating new products sold on a multinational scale. Through registries, drug regulators ensure transparency and dissemination of evaluation methods and endpoint setting. The FDA is a prime example of drug regulation and offers inspiration for how complex AI systems like foundation models could be governed.
Methodology and scope
This report draws on expert interviews and literature to examine the suitability of applying FDA oversight mechanisms to foundation models. It includes lessons drawn from a literature review[38] [39] and interviews with 20 experts from industry, academia, thinktanks and government on FDA oversight and foundation model evaluation processes.[40] In this paper, we answer two core research questions:
- Under what conditions are FDA-style pre-market approval mechanisms successful in reducing risks for drug development and medical software?
- How might these mechanisms be applied to the governance of foundation models?
The report is focused on the applicability of aspects of FDA-style oversight (such as pre-approvals) to foundation models for regulation within a specific jurisdiction. It does not aim to determine if the FDA’s approach is the best for foundation model governance, but to inform policymakers’ decision-making. This report also does not answer how the FDA should regulate foundation models in the medical context.[41]
We focus on how foundation models might be governed within a jurisdiction, not on international cross-jurisdiction oversight. An international approach could be built on top of jurisdictional FDA-style oversight models through mutual recognition and trade limitations, as recently proposed.[42] [43]
We focus particularly on auditing and approval mechanisms, outlining criteria relevant for a future comparative analysis with other national and multinational regulatory models. Further research is needed to understand whether a new agency like the FDA should be set up for AI.
The implications and recommendations of this report will apply differently to different jurisdictions. For example, many downstream ‘high-risk’ applications of foundation models would have the equivalent of a regulatory approval gate under the EU AI Act (due to be finalised at the end of 2023). The most relevant learnings for the EU would therefore be considerations of what upstream foundation model approval gates could entail, or how a post-market monitoring regime should operate. For the UK and USA (and other jurisdictions), there may be more scope to glean ideas about how to implement an FDA-style regulatory framework to cover the whole foundation model supply chain.
‘The FDA oversight process’ chapter explores how FDA oversight functions and its strengths and weaknesses as an approach to risk reduction. We use Software as a Medical Device (SaMD) as a case study to examine how the FDA approaches the regulation of current ‘narrow’ AI systems (AI systems that do not have general capabilities). Then, the chapter on ‘FDA-style oversight for foundation models’ explores the suitability of this approach to foundation models. The paper concludes with recommendations for policymakers and open questions for further research.
Definitions
● Approval gates are the specific points in the FDA oversight process at which regulatory approval decisions are made. They are throughout the development process. A gate can only be passed when the regulator believes that sufficient evidence on safety and efficacy has been provided.
● Class I–III medical devices: Class I medical devices are low-risk with non-critical consequences. Class II devices are medium risk. Class III devices are devices which can potentially cause severe harms.
● Clinical trials, ‘also known as clinical studies, test potential treatments in human volunteers to see whether they should be approved for wider use in the general population’.[44]
● Endpoints are targeted outcomes of a clinical trial that are statistically analysed to help determine efficacy and safety. They may include clinical outcome assessments or other measures to predict efficacy and safety. The FDA and developers jointly agree on endpoints before a clinical trial.
● Foundation models are ‘AI models capable of a wide range of possible tasks and applications, such as text, image, or audio generation. They can be standalone systems or can be used as a ‘base’ for many other more narrow AI applications’.[45]
○ Upstream (in the foundation model supply chain) refers to the component parts and activities in the supply chain that feed into development of the model.[46]
○ Downstream (in the foundation model supply chain) refers to activities after the launch of the model and activities that build on a model.[47]
○ Fine-tuning is the process of training a pre-trained model with an additional specialised or context-specific dataset, removing the need to train a model from scratch.[48]
● Narrow AI is ‘designed to be used for a specific purpose and is not designed to be used beyond its original purpose’.[49]
● Pre-market approval is the point in the regulatory approval process where developers provide evidence on the safety risks, efficacy and accessibility of their products before they are approved to be sold in a market. Beyond pre-market, the term ‘pre-approvals’ generally describes a regulatory approval process before the next step along the development process or supply chain.
● A Quality Management System (QMS) is a collection of business processes focused on achieving quality policy and objectives to meet requirements (see, for example ISO 9001 and ISO 13485),[50] [51] or on safety and efficacy (see, for example FDA Part 820). This includes management controls; design controls; production and process controls; corrective and preventative actions; material controls; records, documents, and change controls; and facilities and equipment controls.
● Risk-based regulation ‘focuses on outcomes rather than specific rules and process as the goal of regulation’,[52] adjusting oversight mechanisms to the level of risk of the specific product or technology.
● Software as a Medical Device (SaMD) is ‘Software intended to be used for one or more medical purposes that perform these purposes without being part of a hardware medical device’.[53]
● The US Food and Drug Administration (FDA) is a federal agency (and part of the Department of Health and Human Services) that is charged with protecting consumers against impure and unsafe foods, drugs and cosmetics. It enforces the Federal Food Drug and Cosmetic Act and related laws, and develops detailed guidelines.
How to read this paper
This report offers insight from FDA regulators, civil society and private sector companies on applying specific oversight mechanisms proven in life sciences, to govern AI and foundation models specifically.
…if you are a policymaker working on AI regulation and oversight:
- The section on ‘Applying key features of FDA-style oversight to foundation models’ provides general principles that can contribute to a risk-reducing approach to oversight,
- The chapter on ‘Recommendations and open questions’ summarises specific mechanisms for developing and implementing oversight for foundation models.
- For a detailed analysis of the applicability of life sciences oversight to foundation models, see the chapter ‘FDA-style oversight for foundation models’ and section on ‘The limitations of FDA oversight’.
…if you are a developer or designer of data-driven technologies, foundation models or AI systems:
- Grasp the importance of rigorous testing, documentation and post-market monitoring of foundation models and AI applications. The introduction and ‘FDA-style oversight for foundation models’ chapter detail why significant investments into AI governance is important, and why the life sciences are a suitable inspiration.
- The section on ‘Applying specific FDA-style processes along the foundation model supply chain’ describes mechanisms for each layer in the foundation model supply chain, They are tailored to data providers, foundation model developers, hosts and application providers. These mechanisms are based on proven governance methods used by regulators and companies in the pharmaceutical and medical device sectors.
- Our Recommendations and open questions’ provide actionable ways in which AI companies can contribute to a better AI oversight process.
…if you are a researcher or public engagement practitioner interested in AI regulation:
- The introduction includes an overview of the methodology which may also offer insight for others interested in undertaking a similar research project.
- In addition to a summary of the FDA oversight process, the main research contribution of this paper is in the chapter ‘FDA-style oversight for foundation models’.
- Our chapter on ‘Recommendations and open questions’ outlines opportunities for future research on governance processes.
- There is also potential in collaborations between researchers in life sciences regulation and AI governance, focusing on the specific oversight mechanisms and technical tools like unique device identifiers described in our recommendations for AI regulators, developers and deployers.
The FDA oversight process
The Food and Drug Administration (FDA) is the US federal agency tasked with enforcing laws on food and drug products. Its core objective is to help ‘speed innovations that make products more effective, safer and more affordable’ through ‘accurate, science-based information’. In 2023, it had a budget of around $8 billion, around half of which was paid through mandatory fees by companies overseen by the FDA.[54] [55]
The FDA’s regulatory mandate has come to include regulating computing hardware and software used for medical purposes, such as in-vitro glucose monitoring devices or breast cancer diagnosis software.[56] The regulatory category SaMD and adjacent software for medical devices encompasses AI-powered medical applications. These are novel software applications that may bear potentially severe consequences, such as software for eye surgeries[57] or automated oxygen level control under anaesthesia.[58] [59]
An understanding of the most important oversight components for the FDA enables the discussion on suitable inspirations for foundation models in the following chapter.
The FDA regulates drugs and medical devices through a risk-based approach. This seeks to identify potential risks at different stages of the development process. The FDA does this by providing guidance and setting requirements for drug and device developers, including agreed protocols for testing and evaluating the safety and efficacy of the drug or device. The definition of ‘safety’ and ‘efficacy’ are dependent on the context, but generally:
- Safety refers to the type and likelihood of adverse effects. This is then described as ‘a judgement of the acceptability of the risk associated with a medical technology’. A ‘safe’ technology is described as one that ‘causes no undue harm’.[60]
- Efficacy refers to ‘the probability of benefit to individuals in a defined population from a medical technology applied for a given medical problem’.[61] [62]
Some devices and drugs undergo greater scrutiny than others. For medical devices, the FDA has developed a Class I–III risk rating system; higher-risk (Class III) devices are required to meet more stringent requirements to be approved and sold on the market. For medical software, the focus lies more on post-market monitoring. The FDA allows software on the market with higher levels of risk uncertainty than drugs, but it monitors such software continuously.
Figure 3: Classes of medical devices (applicable to software components and SaMD)[63]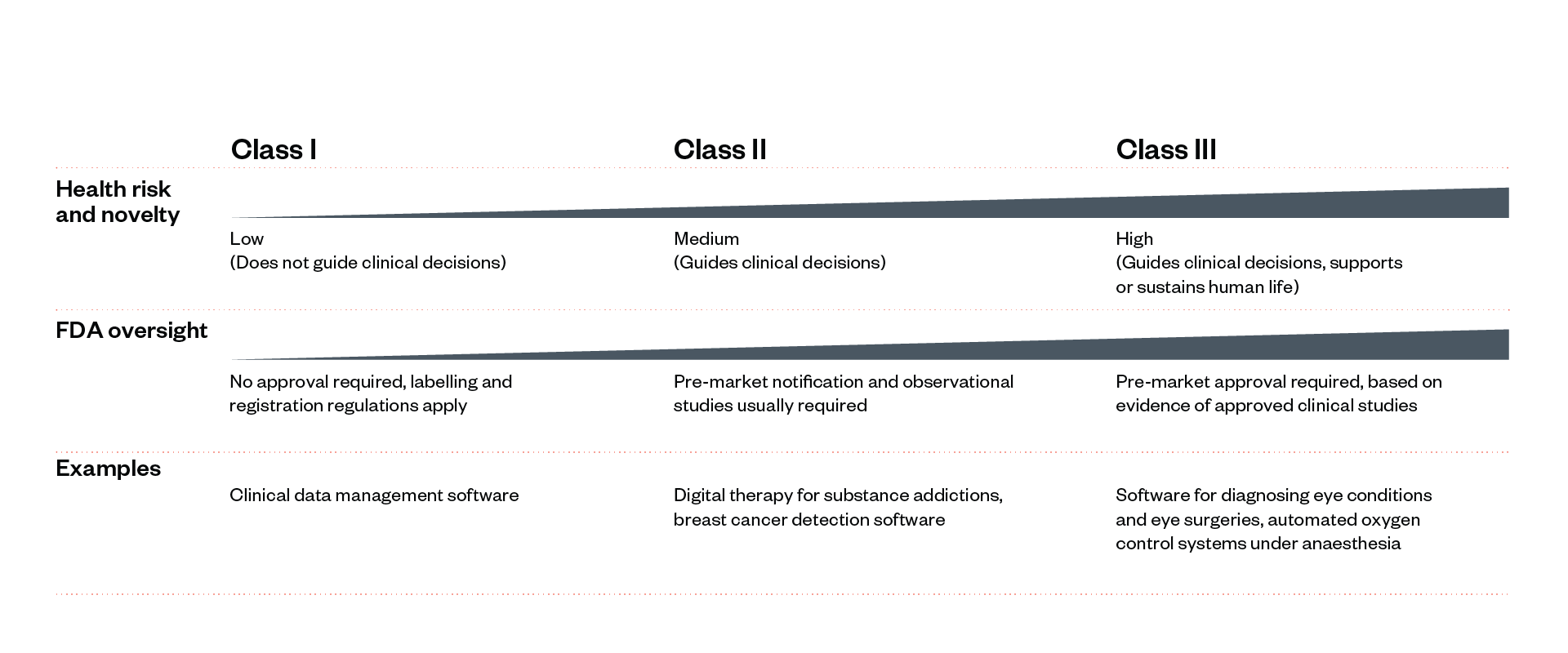
The FDA’s oversight process follows five steps, which are adapted to the category and risk class of the drug, software or medical device in question.[64] [65]
The FDA can initiate reviews and inspections of drugs and medical devices (as well as other medical and food products) at three points: before clinical trials begin (Step 2), before a drug is marketed to the public (Step 4) and as part of post-market monitoring (Step 5). The depth of evidence required depends on the potential risk levels and novelty of a drug or device.
Approval gates – points in the development process where proof of sufficient safety and efficacy is required to move to the next step – are determined depending on where risks originate and proliferate.
This section illustrates the FDA’s oversight approach to novel Class III software (including narrow AI applications). Low-risk software and software similar to existing software go through a significantly shorter process (see Figure 3).
We illustrate each step using the hypothetical scenario of an approval process for medical AI software for guiding a robotic arm to take patients’ blood. This software consists of a neural network that has been trained with an image classification dataset to visually detect an appropriate vein and that can direct a human or robotic arm to this vein (see Figure 4).[66] [67]
While the oversight process for drugs and medical devices is slightly different, this section borrows insights from both and simplifies when suitable. This illustration will help to inform our assessment in the following chapter, of whether and how a similar approach could be applied to ensure oversight of foundation models.
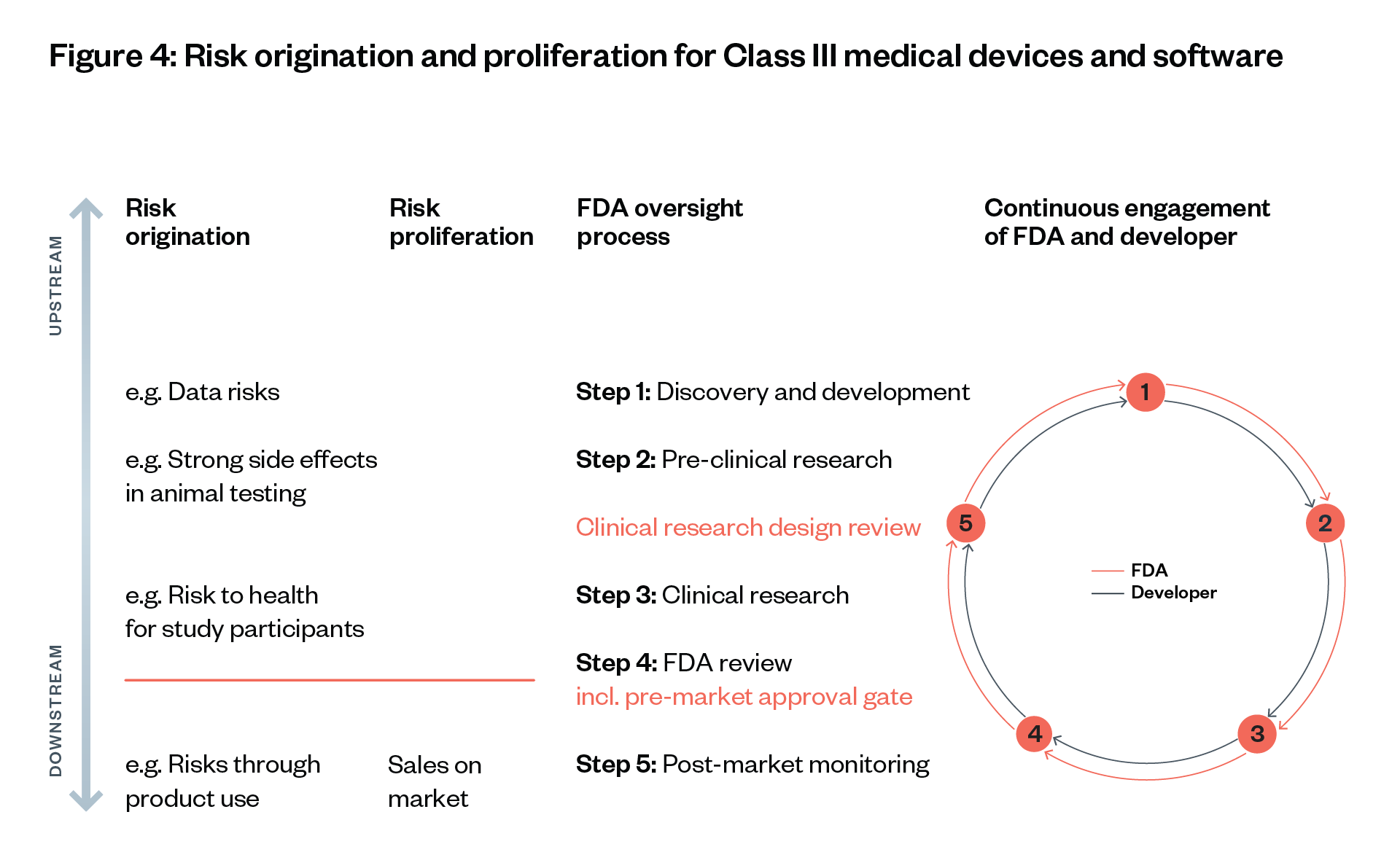
Risk origination points are when risks arise initially; risk proliferation points: when risks spread without being controllable any more.
Step 1: Discovery and development
Description: A developer conducts initial scoping and ideation of how to design a medical device or drug, including use cases for the new product, supply chain considerations, regulatory implications and needs of downstream users. At the start of the development process, the FDA uses pre-submissions, which aim to provide a path from conceptualisation through to placement on the market.
Developer responsibilities:
- Determine the product and risk category to classify the device, which will determine the testing and evaluation procedure (see Figure 3).
- While training the AI model, conduct internal (non-clinical) tests, and clearly document the data and algorithms used throughout the process in a Quality Management System (QMS).[68]
- Follow Good Documentation Practice, which offer guidance on how to document procedures from development through to market, to facilitate risk mitigation, validation and verification, and traceability (to support regulators in the event of recall or investigations).
- Inform the FDA on the necessity of new software, for example, for efficiency gains or improvements in quality.
FDA responsibilities:
- Support developers in risk determination.
- Offer guidance on, for example, milestones for (pre-)clinical research and data analysis.
Required outcomes: Selection of product and risk category to determine regulatory pathway.
Example scenario: A device that uses software to guide the taking of blood may be classified as an in-vitro diagnostics device, which the FDA has previously classified as Class III (highest risk class).[69]
Step 2: Pre-clinical research
Description: In this step, basic questions about safety are addressed through initial animal testing.
Developer responsibilities:
- Propose endpoints of study and conduct research (often with a second party).
- Use continuous tracking in the QMS and share results with FDA.
FDA responsibilities:
- Approve endpoints of the study, depending on the novelty and type of medical device or drug.
- Review results to allow progression to clinical research.
Required outcomes: Developer proves basic safety of product, allowing clinical studies with human volunteers in the next step.
Example scenario: This step is important for assessing risks of novel drugs. It would not usually be needed for medical software such as our example that helps take blood, as these types of software are typically aimed at automating or improving existing procedures.
Step 3: Clinical research
Description: Drugs, devices and software are tested on humans to make sure they are safe and effective. Unlike for foundation models and most AI research and development, institutional review for research with human subjects is mandatory in public health.
Developer responsibilities:
- Create a research design (called a protocol) and submit it to an institutional review board (IRB) for ethical review, along with Good Clinical Practice (GCP) principles and ISO standards such as ISO14155.
- Provide the FDA with the research protocol, the hypotheses and results of the clinical trials and of any other pre-clinical or human tests undertaken, and other relevant information.
- Following FDA approval, hire an independent contractor to conduct clinical studies (as required by risk level); these may be in multiple regions or locations, as agreed with the FDA, to match future application environments.
For drugs, trials may take place in phases that seek to identify different aspects of a drug:
- Phase 1 studies tend to involve less than 100 participants, run for several months and seek to identify the safety and dosage of a drug.
- Phase 2 studies tend to involve up to several hundred people with the disease/condition, run for up to two years and study the efficacy and side effects.
- Phase 3 studies involve up to 3,000 volunteers, can run for one to four years and study efficacy and adverse reactions.
FDA responsibilities:
- Approve the clinical research design protocol before trials can proceed.
- During testing, support the developer with guidance or advice at set intervals on protocol design and open questions.
Required outcomes: Once the trials are completed, the developer submits them as evidence to the FDA. The supplied information should include:
- description of main functions
- data from trials to prove safety and efficacy
- benefit/risk and mitigation review, citing relevant literature and medical association guidelines
- intended use cases and limitations
- a predetermined change control plan, allowing for post-approval adaptations of software without the need for re-approval (for a new use, new approval is required)
- QMS review (code, protocols of storing data, Health Protection Agency guidelines, patient confidentiality).
Example scenario: The developers submit a ‘submission of investigational device exemption’ to the FDA, seeking to simplify design, requesting observational studies of the device instead of randomised controlled trials. They provide a proposed research design protocol to the FDA. Once the FDA approves it, they begin trials in 15 facilities with 50 patients each, aiming to prove 98 per cent accuracy and reduction of waiting times at clinics. During testing, no significant adverse events are reported. The safety and efficacy information is submitted to the FDA.
Step 4: FDA review
Description: FDA review teams thoroughly examine the submitted data on the drug or device and decide whether to approve it.
Developer responsibilities: Work closely with the FDA to provide access to all requested information and facilities (as described above).
FDA responsibilities:
- Assign specialised staff to review all submitted data.
- In some cases, conduct inspections and audits of developer’s records and evidence, including site visits.
- If needed, seek advice from an advisory committee, usually appointed by the FDA Commissioner with input from the federal Secretary of the Health & Human Service department.[70] The committee may include representation from patients, scientific academia, consumer organisations and industry (if decision-making is delegated to the committee, only scientifically qualified members may vote).
Required outcomes: Approval and registration, or no approval with request for additional evidence.
Example scenario: For novel software like the example here, there might be significant uncertainty. The FDA could request more information from the developer and consult additional experts. Decision-making may be delegated to an advisory committee to discuss open questions and approval.
Step 5: Post-market monitoring
Description: The aim of this step is to detect ‘adverse events’[71] (discussed further below) to increase safety iteratively. At this point, all devices are labelled with Unique Device Identifiers to support monitoring and reporting from development through to market. These are particularly in relation to identifying the underlying causes of, and corrective actions for adverse events.
Developer responsibilities: Any changes or upgrades must be clearly documented, within the agreed change control plan.
FDA responsibilities:
- Monitor safety of all drugs and devices once available for use by the public.
- Monitor compliance on an ongoing basis through the QMS, with safety and efficacy data reviewed every six to 12 months.
- Maintain a database on adverse events and recalls.[72]
Required outcomes: No adverse events or diminishing efficacy. If safety issues occur, the FDA may issue a recall.
Example scenario: Due to a reported safety incident with the blood-taking software, the FDA inspects internal emails and facilities. In addition, every six months, the FDA reviews a one per cent sample of patient data in the QMS and conducts interviews with patients and staff from a randomly selected facility.
Risk-reducing aspects of FDA oversight
Our interviews with experts on the FDA and a literature review[73] highlighted several themes. We group them into five risk-reducing aspects below.
Risk- and novelty-driven oversight
The approval gates described in the previous section lead to iterative oversight using QMS and jointly agreed research endpoints, as well as continuous post-market monitoring.
Approval gates are informed by risk controllability. Risk controllability is understood by considering the severity of harm to people; the likelihood of that harm occurring; proliferation, duration of exposure to population; potential false results; patient tolerance of risk; risk factors for people administering or using the drug or device, such as caregivers; detectability of risks; risk mitigations; the drug or device developer’s compliance history; and how much uncertainty there may be around any of these factors.[74]
Class III devices and related software – those that may guide critical clinical decisions or that are invasive or life-supporting – need FDA pre-approval before the drug is marketed to the public. In addition, the clinical research design needs to be approved by the FDA.
Continuous, direct engagement of FDA with developers throughout the development process
There can be inspections at any step of the development and deployment process. Across all oversight steps, the FDA’s assessments are independent and not reliant on input from private auditors who may have profit incentives.
In the context of foundation models, where safety standards are unclear and risk assessments are therefore more exploratory, these assessments should not be guided by profit incentives.
In cases where the risks are less severe, for example, Class II devices, the FDA is supported by accredited external reviewers.[75] External experts also support reviews of novel technology where the FDA lacks expertise, although this approach has been criticised (see limitations below and advisory committee description above).
FDA employees review planned clinical trials, as well as clinical trial data produced by developers and their contractors. In novel, high-stakes cases, a dedicated advisory committee reviews evidence and decides on approval. Post market, the FDA reviews sample usage, complaint and other data approximately every six months.
Wide-ranging information access
By law, the FDA is empowered to request comprehensive evidence through audits, conduct inspections[76] and check the QMS. The FDA’s QMS regulation requires documented, comprehensive managerial processes for quality planning, purchasing, acceptance activities, nonconformities and corrective/preventative actions throughout design, production, distribution and post-market. While the FDA has statutory powers to access comprehensive information, for example, on clinical trials, patient data and in some cases internal emails, it releases only a summary of safety and efficacy post approval.
Putting the burden of proof on the developer
The FDA must approve clinical trials and their endpoints, and the labelling materials for drugs and medical devices, before they are approved for market. This model puts the burden of proof on the developer to provide this information or be unable to sell their product.
A clear approval gate entails the following steps:
- The product development process in scope: The FDA’s move into regulating SaMD required it to translate regulatory approval gates for a drug approval process to the stages of a software development process. In the SaMD context, a device may be made up of different components, including software and hardware, that come from other suppliers or actors further upstream in the product development process. The FDA ensures the safety and efficacy of each component by requiring all components to undergo testing. If a component has been previously reviewed by the FDA, future uses of it can undergo an expedited review. In some cases, devices may use open-source Software of Unknown Provenance (SOUP). Such software needs either to be clearly isolated from critical components of the device, or to undergo demonstrable safety testing.[77]
- The point of approval in the product development process: Effective gates occur once a risk is identifiable, but before it can proliferate or turn into harms. Certain risks (such as differential impacts on diverse demographic groups) may not be identifiable until after the intended uses of the device are made clear (for example will it be used in a hospital or a care home?). For technology with a wide spectrum of uses, like gene editing, developers must specify intended uses and the FDA allows trials with human subjects only in a few cases, where other treatments have higher risks or significantly lower chance of success.[78]
- The evidence required to pass the approval gate: This is tiered depending on the risk class, as already described. The FDA begins with an initial broad criterion such as simply not causing to the human body when used. Developers and contractors then provide exploratory evidence. Based on this, in the case of medicines, the regulator learns and makes further specifications, for example, around the drug elimination period. For medical devices such as heart stents, evidence could include the percentage reduction in the rate of major cardiac events.
Balancing innovation and risks enables regulatory authority to be built over time
The FDA enables innovation and access by streamlining approval processes (for example, similarity exemptions, pre-submissions) and approvals of drugs with severe risks but high benefits. Over time, Congress has provided the FDA with increasing information access and enforcement powers and budgets, to allow it to enforce ‘safe access’.
The FDA has covered more and more areas over time, recently adding tobacco control to its remit.[79] FDA-regulated products account for about 20 cents of every dollar spent by US consumers.[80] It has the statutory power to issue warnings, make seizures, impose fines and pursue criminal prosecution.
Safety and accessibility need to be balanced. For example, a piece of software that automates oxygen control may perform slightly less well than healthcare professionals, but if it reduces the human time and effort involved and therefore increases accessibility, it may still be beneficial overall. By finding the right balance, the FDA builds an overall reputation as an agency providing mostly safe access, enabling continued regulatory power.[81] When risk uncertainty is high, it can slow down the marketing of technologies, for example, allowing only initial, narrow experiments for novel technologies such as gene editing.[82]
The FDA approach does not rely on any one of these risk-reducing aspects alone. Rather, the combination of all five ensures the safety of FDA-regulated medical devices and drugs in most cases.[83] The five together also allow the FDA to continuously learn about risks and improve its approval process and its guidance on safety standards.
Risk- and novelty-driven oversight focuses learning on the most complex and important drugs, software and devices. Direct engagement and access to a wide range of information is the basis of the FDA’s understanding of new products and new risks.
With the burden of proof on developers through pre-approvals, they are incentivised to ensure the FDA is informed about safety and efficacy.
As a result of this approach to oversight, the FDA is better able to balance safety and accessibility, leading to increased regulatory authority.
‘The burden is on the industry to demonstrate the safety and effectiveness, so there is interest in educating the FDA about the technology.’
Former FDA Chief Counsel
The history of the FDA: 100+ years of learning and increasing power [84] [85] [86]
The creation of the FDA was driven by a series of medical accidents that exposed the risks drug development can pose to public safety. While the early drug industry initially pledged to self-regulate, and members of the public viewed doctors as the primary keepers of public safety, public outcry over tragedies like the Elixir Sulfanilamide disaster (see below) led to calls for an increasingly powerful federal agency.
Today the FDA employs around 18,000 people (2022 figures) with a $8 billion budget (2023 data). The FDA’s approach to regulating drugs and devices involves learning iteratively about risks and benefits of products with every new evidence review it undertakes as part of the approval process.
Initiation
The 1906 Pure Food and Drugs Act was the first piece of legislation to regulate drugs in the USA. A groundbreaking law, it took nearly a quarter-century to formulate. It prohibited interstate commerce of adulterated and misbranded food and drugs, marking the start of federal consumer protection.
Learning through trade controls: This Act established the importance of regulatory oversight for product integrity and consumer protection.
Limited mandate
From 1930 to 1937, there were failed attempts to expand FDA powers, with relevant bills not being passed by Congress. This period underscored the challenges in evolving regulatory frameworks to meet public health needs.
Limited power and limited learning.
Elixir Sulfanilamide disaster
This 1937 event, where an untested toxic solvent caused over 100 deaths, marked a turning point in drug safety awareness.
Learning through post-market complaints: The Elixir tragedy emphasised the crucial need for pre-market regulatory oversight in pharmaceuticals.
Extended mandate
In 1938, previously proposed legislation, the Food, Drug, and Cosmetic Act, was passed into law that changed the FDA’s regulatory approach by mandating review processes without requiring proof of fraudulent intent.
Learning through mandated information access and approval power: Pre-market approvals and the FDA’s access to drug testing information enabled the building of appropriate safety controls.
Safety reputation
During the 1960s, the FDA’s refusal to approve thalidomide –a drug prescribed to pregnant women causing an estimated 80,000 miscarriages and infant deaths and deformities in 20,000 children worldwide – further established its commitment to drug safety.
Learning through prevented negative outcomes: The thalidomide situation led the FDA to calibrate its safety measures by monitoring and preventing large-scale health catastrophes, especially in comparison with similar countries. Post-market recalls were included in the FDA’s regulatory powers.
Extended enforcement power
The 1962 Kefauver-Harris Amendment to the Federal Food, Drug, and Cosmetic Act was a significant step, requiring new drug applications to provide substantial evidence of efficacy and safety.
Learning through expanded enforcement powers: This period reinforced the evolving role of drug developers in demonstrating the safety and efficacy of their products.
Balancing accessibility with safety
The 1984 Drug Price Competition and Patent Term Restoration Act marked a balance between drug safety and accessibility, simplifying generic drug approvals. In the 2000s, Risk Minimization Action Plans were introduced, emphasising the need for drugs to have more benefits than risks, monitored at both the pre- and the post-market stages.
Learning through a lifecycle approach: This era saw the FDA expanding its oversight scope across product development and deployment for a deeper understanding of the benefit–risk trade-off.
Extended independence
The restructuring of advisory committees in the 2000s and 2010s enhanced the FDA’s independence and decision-making capability.
Learning through independent multi-stakeholder advice: The multiple perspectives of diverse expert groups bolstered the FDA’s ability to make well-informed, less biased decisions, reflecting a broad range of scientific and medical insights – although critics and limitations remain (see below).
Extension to new technologies
In the 2010s and 2020s, recognising the potential of technological advancements to improve healthcare quality and cost efficiency, the FDA began regulating new technologies such as AI in medical devices.
Learning through a focus on innovation: Keeping an eye on emerging technologies.
The limitations of FDA oversight
The FDA’s oversight regime is built for regulating food, drugs and medical devices, and more recently extended to software used in medical applications. Literature reviews[87] and interviewed FDA experts suggest three significant limitations of this regime’s applicability to other sectors.
Limited types of risks controlled
The FDA focuses on risks to life posed by product use, therefore focusing on reliability and (accidental) misuse risks. Systemic risks such as accessibility challenges, structural discrimination issues and novel risk profiles are not as well covered.[88] [89]
- Accessibility risks include the cost barriers of advanced biotechnology drugs or SaMD for underprivileged groups.[90]
- Structural discrimination risks include disproportionate risks to particular demographics caused by wider societal inequalities and a lack of representation in data. These may not appear in clinical trials or in single-device post-market monitoring. For example, SaMD algorithms have misclassified Black patients’ healthcare needs systematically because they have suggested treatment based past healthcare spending data that did not accurately reflect their requirements.[91]
- Equity risks arise when manufacturers claim average accuracy across a population or use only for a specific population (for example, people aged 60+). The FDA only considers whether a product safely and effectively delivers according to the claims of its manufacturers – it doesn’t go beyond this to urge them to reach other populations. It does not yet have comprehensive algorithmic impact assessments to ensure equity and fairness.
- False similarity risks originate in the accelerated FDA 510(k) approval pathway for medical devices and software through comparison with already-approved products –referred to as predicate devices. Reviews of this pathway have shown ‘predicate creep’ when multiple generations of predicate devices slowly drift away from the originally approved use.[92] This could mean that predicate devices may not provide suitable comparisons for new devices.
- Novel risk profiles challenge the standard regulatory approach of the FDA that rests on risk detection through trials before risks proliferate through marketing. Risks that are not typically detectable in clinical trials, due to their novelty or new application environments, may be missed. For example, the risk of water-contaminating foods is clear, but it may be less clear how to monitor for new pathogens that might be significantly smaller or otherwise different to those detected by existing routines.[93] While any ‘adverse events’ need to be reported to the FDA, risks that are difficult to detect might be missed.
Limited number of developers due to high costs of compliance
The FDA’s stringent approval requirements lead to costly approval processes that only large corporations can afford, as a multi-stage clinical trial can cost tens of millions of dollars.[94] [95] This can lead to oligopolies and monopolies, high drug prices because of limited competition, and innovation focused on areas with high monetary returns.
If this is not counteracted through governmental subsidies and reimbursement incentives, groups with limited means to pay for medications can face accessibility issues. It remains an open question whether small companies should be able to develop and market severe-risk technologies, or how governmental incentives and efforts can democratise the drug and medical device – or foundation model – development process.
Reliance on industry for expertise
The FDA sometimes relies on industry expertise, particularly in novel areas where clear benchmarks have not been developed and knowledge is concentrated in industry. This means that the FDA may seek input from external consultants and its advisory committees to make informed decisions.[96]
An overreliance on industry could raise concerns around regulatory capture and conflicts of interest – similar to other agencies.[97] For example, around 25 per cent of FDA advisory committee members had conflicts of interest in the past five years.[98] In principle, conflicted members are not allowed to participate, but dependency on their expertise regularly leads this requirement being waived.[99] [100] [101] External consultants have been conflicted, too: one notable scandal occurred when McKinsey advised the FDA on opioid policy while being paid by corporations to help them sell the same drugs.[102]
A lack of independent expertise can reduce opportunities for the voice of people affected by high-risk drugs or devices being heard. This in turn may undermine public trust in new drugs and devices. It has also been shown that oversight processes that are not heavily dependent on industry expertise and funding have been proven to discover more, and more significant, risks and inaccuracies.[103]
Besides these three main limitations, others include enforcement issues for small-scale illegal deployment of SaMD, which can be hard to identify;[104] [105] and device misclassifications in new areas.[106]
FDA-style oversight for foundation models
FDA Class III devices are complex, novel technologies with potentially severe risks to public health and uncertainties regarding how to detect and mitigate these risks.[107]
Foundation models are at least as complex, more novel and – alongside their potential benefits – likewise pose potentially severe risks, according to the experts we interviewed and recent literature.[108] [109] [110] They are also deployed across the economy, interacting with millions of people, meaning they are likely to pose systemic risks that are far beyond those of Class III medical devices.[111]
However, the risks of foundation models are so far not fully clear, risk mitigation measures are uncertain and risk modelling is poor or non-existent.
Leading AI researchers such as Stuart Russell and Yoshua Bengio, independent research organisations, and AI developers have flagged the riskiness, complexity and black-box nature of foundation models.[112] [113] [114] [115] [116] In a review on the severe risks of foundation models (in this case, the accessibility of instructions for responding to biological threats), the AI lab Anthropic states: ‘If unmitigated, we worry that these risks are near-term, meaning they may be actualised in the next two to three years.’[117]
As seen in the history of the FDA outlined above, it was a reaction to severe harm that led to its regulatory capacity being strengthened. Those responsible for AI governance would be well advised to act ahead of time to pre-empt and reduce the risk of similarly severe harms.
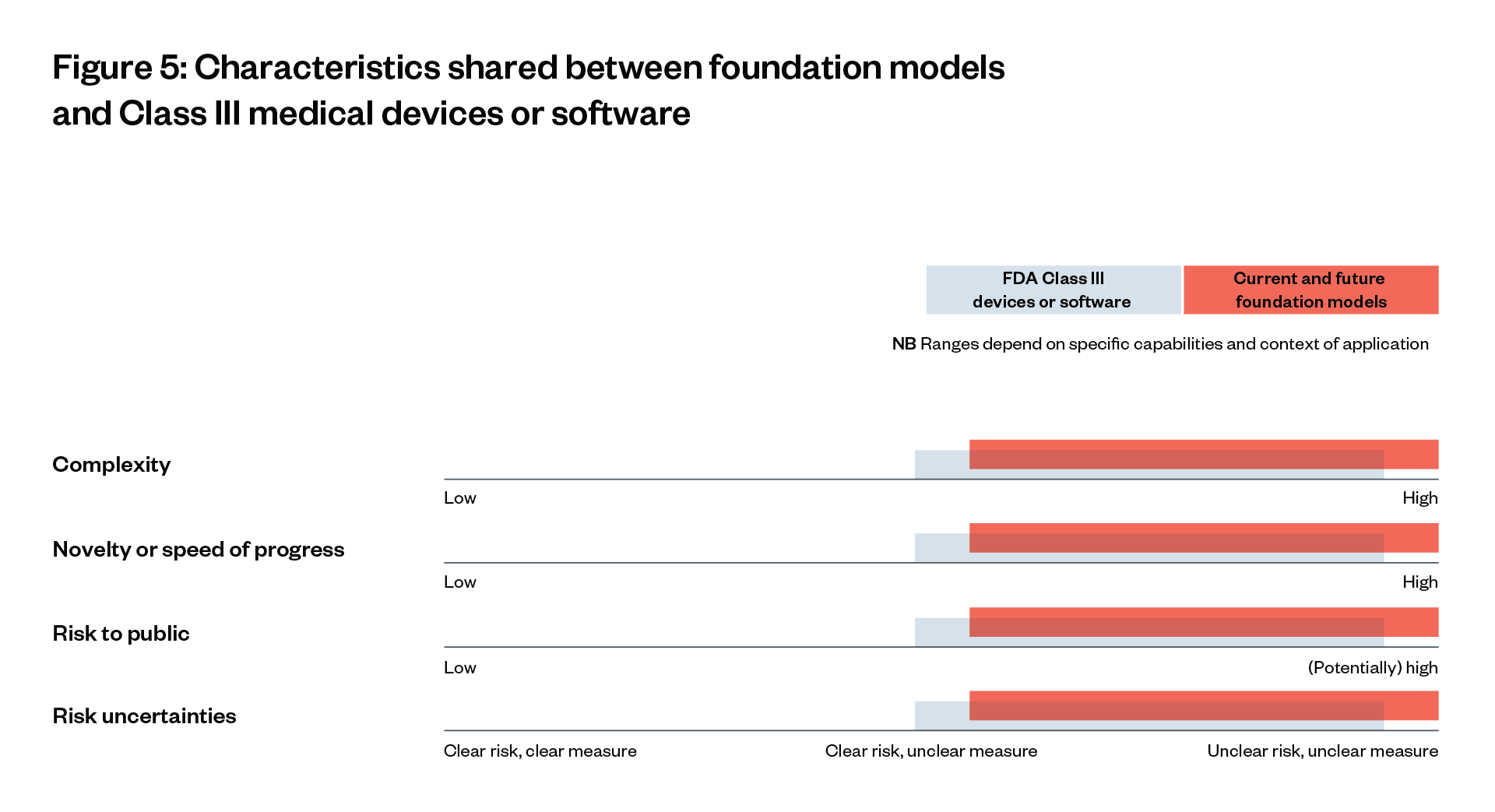
The similarities between foundation models and existing, highly regulated Class III medical devices – in terms of complexity, novelty and risk uncertainties – suggests that they should be regulated in a similar way (see Figure 5).
However, foundation models differ in important ways from Software as a Medical Device (SaMD). The definitions themselves reveal inherent differences in the range of applications and intended use:
Foundation models are AI models capable of a wide range of possible tasks and applications, such as text, image or audio generation. They can be stand-alone systems or can be used as a ‘base’ for many other more narrow AI applications.[118]
SaMD is more specific: it is software that is ‘intended to be used for one or more medical purposes that perform[s] these purposes without being part of a hardware medical device’.[119]
However, the most notable differences are more subtle. Even technology applied across a wide range of purposes, like general drug dispersion software, can be effectively regulated with pre-approvals. This is because the points of risk and the pathways to dangerous outcomes are well understood and agreed upon, and they all start from the distribution of products to consumers – something in which the FDA can intervene.
The first section of this chapter outlines why this is not yet the case for foundation models. The second section illustrates how FDA-style oversight can bridge this gap generally. The third section details how these mechanisms could be applied along the foundation model supply chain – the different stages of development and deployment of these models.
The foundation model challenge: unclear, distributed points of risk
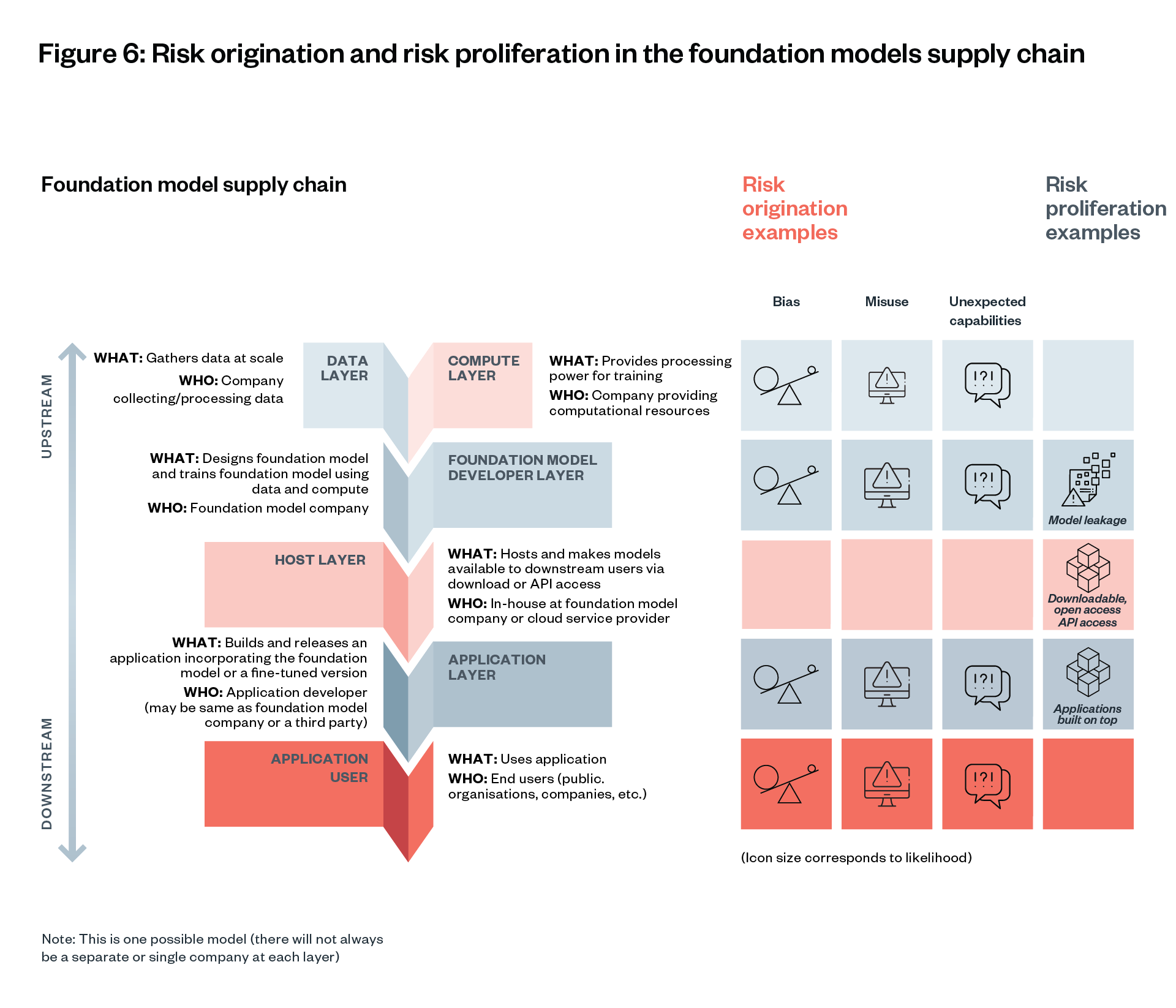
In this section we discuss two key points of risk: 1) risk origination points, when risks arise initially; and 2) risk proliferation points, when risks spread without being controllable.
A significant challenge that foundation models raise is the difficulty of identifying where different risks originate and proliferate in their development and deployment, and which actors within that process should be held responsible for mitigating and providing redress for those harms.[120]
Risk origination and proliferation examples
Bias
Some risks may originate in multiple places in the foundation model supply chain. For example, the risk of a model producing outputs that reinforce racial stereotypes may originate in the data used to train the model, how it was cleaned, the weights that the model developer used, which users the model was made available to, and what kinds of prompts the end user of the model is allowed to make.[121] [122]
In this example, a series of evaluations for different bias issues might be needed throughout the model’s supply chain. The model developer and dataset provider would need to be obliged to proactively look for and address known issues of bias. It might also be necessary to find ways to prohibit or discourage end users from prompting a model for outputs that reinforce racial stereotypes.
Cybercrime
Another example is reports of GPT-4 being used to write code for phishing operations to steal people’s personal information. Where in the supply chain did such cyber-capabilities originate and proliferate?[123] [124] Did the risk originate during training (while general code-writing abilities were being built) or after release (allowing requests compatible with phishing)? Did it proliferate through model leakage, widely accessible chatbots like ChatGPT or Application Programming Interfaces (APIs), or downstream applications?
Some AI researchers have conceptualised the uncertainty over risks as a matter of the unexpected capabilities of foundation models. This ‘unexpected capabilities problem’ may arise during models’ development and deployment.[125] Exactly what risks this will lead to cannot be identified reliably, especially not before the range of potential use cases is clear.[126] In turn, this uncertainty means that risks may be more likely to proliferate rapidly (the ‘proliferation problem’),[127] and to lead to harms throughout the lifecycle – with limited possibility for recall (the ‘deployment safety problem’).[128]
The challenge in governing foundation models is therefore in identifying and mitigating risks comprehensively before they proliferate.[129]
There is a distinction to draw between risk origination (the point in the supply chain a risk such as toxic content may arise) and risk proliferation (the point in the supply chain a risk can be widely distributed to downstream actors). Identifying points of risk origination and proliferation can be challenging for different kinds of risks.
Foundation model oversight needs to be continuous throughout the supply chain. Identifying all inherent risks in a foundation model upstream is hard. Leaving risks to downstream companies is not the solution, because they may have proliferated already by this stage.
There are tools available to help upstream foundation model developers reduce risk before training (through filtering data inputs), and to assess risks during training (through clinical trial style protocols). More of these tools are needed. They are most effective when applied at the foundation model layer (see Figure 2 and Figure 6), given the centralised nature of foundation models. However, some risks might arise or be detectable only at the application layer, so tools for intervention at this layer are also necessary.
Applying key features of FDA-style oversight to foundation models
How should an oversight regime be designed so that it suits complex, novel, severe-risk technologies with distributed, unclear points of risk origination and proliferation?
Both foundation models and Class III devices pose potentially severe levels of risk to public safety and therefore require governmental oversight. For the former, this is arguably even more important given national security concerns (for example, the risk that such technologies could enable cyberattacks or widespread disinformation campaigns at far greater scales than current capabilities allow).[130] [131] [132]
Government oversight is needed also because of the limitations of private insurance for severe risks.
As seen in the cases of nuclear waste insurance or financial crisis, large externalities and systemic risks need to be captured by a government.
Below we consider what we can learn from the oversight of FDA-regulated products and whether an FDA-style approach could provide effective oversight of foundation models.
Building on Raji et al’s recent review[133] and interviews, current oversight regimes for foundation models can be understood alongside, and compared with, the core risk-reducing aspects of the FDA approach, as depicted in Figure 7.[134] [135] Current oversight and evaluations of GPT-4 lag behind FDA oversight in all dimensions.
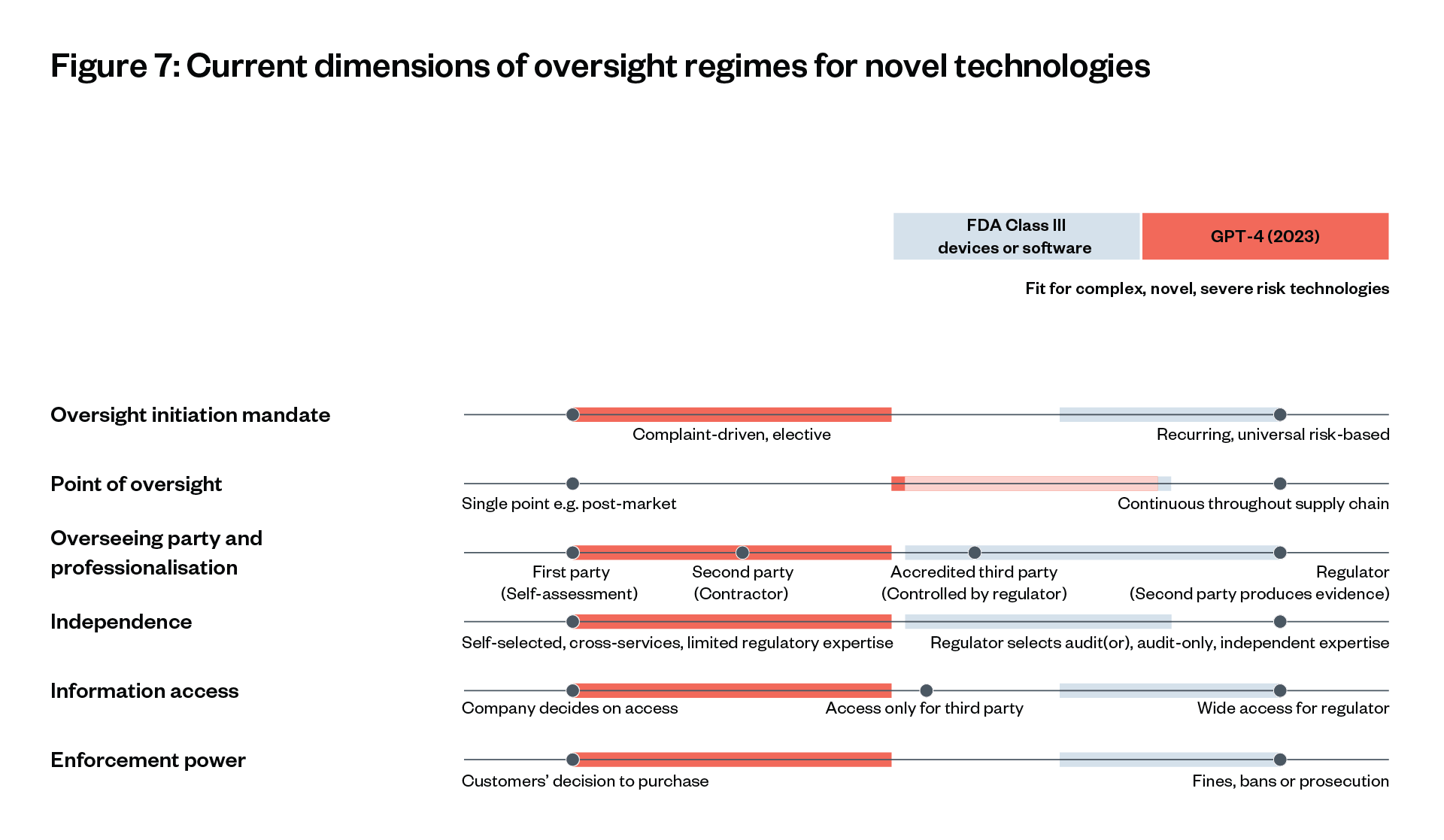
Governance of GPT-4’s development and release according to their 2023 system card and interviews, vs. FDA governance of Class III drugs.[136] [137] [138] While necessarily simplified, characteristics furthest to the right fit best for complex, novel technologies with potentially severe risks and unclear risk (measures).[139]
‘We are in a “YOLO [you only live once]” culture without meaningful specifications and testing – “build, release, see what happens”.’
Igor Krawczuk on current oversight of commercial foundation models
The complexity and risk uncertainties of foundation models could justify similar levels of oversight to those provided by the FDA in relation to Class III medical devices.
This would involve an extensive ecosystem of second-party, third-party and regulatory oversight to monitor and understand the capabilities of foundation models and to detect and mitigate risks. The high speed of progress in foundation model development requires adaptable oversight institutions, including non-governmental organisations with specialised expertise. AI regulators need to establish and enforce improved foundation model oversight across the development and deployment process.
General principles for applying key features of the FDA’s approach to foundation model governance
- Establish continuous, risk-based evaluations and audits throughout the foundation model supply chain. Existing bug bounty programmes[140] and complaint-driven evaluation do not sufficiently cover potential risks. The FDA’s incident reporting system captures fewer risks than the universal risk-based reviews before market entry and post-market monitoring requirements.[141] Therefore, review points need to be defined across the supply chain of foundation models, with risk-based triggers. As already discussed, risks can originate at multiple sources, potentially simultaneously. Continuous engagement of reviewers and evaluators is therefore important to detect and mitigate risks before they proliferate.
- Empower regulatory agencies to evaluate critical safety evidence directly, supported by a third-party ecosystem. First-party self-assessments and second-party contracted auditing have consistently proven to be lower quality than accredited third-party or governmental audits.[142] [143] [144] [145] Regulators of foundation models should therefore have direct access to assess evaluation and audit evidence. This is especially significant when operating in a context when standards are unclear and audits therefore more exploratory (in the style of evaluations). Regulators can also improve their understanding by consulting independent experts.
- Ensure independence of regulators and external evaluators. Oversight processes not dependent on industry expertise and funding have been proven to discover more, and more significant, risks and inaccuracies, especially in complex settings with vague standards.[146] [147] Inspired by the FDA approach, foundation model oversight could be funded directly through mandatory fees from AI labs and only partly through federal funding. Sufficient resourcing in these ways is essential, to avoid the need for additional resourcing that is associated with potential conflicts of interest. Consideration should also be given to an upstream regulator of foundation models as existing sector-specific regulators may only have the ability to review downstream AI applications. The level of funding for such a regulator needs to be similar to that of other safety-critical domains, such as medicine. Civil society and external evaluators could be empowered through access to federal computing infrastructure for evaluations and accreditation programmes.
- Enable structured access to foundation models and adjacent components for evaluators and civil society. Access to information is the foundation of an effective audit (although while it is necessary, it is not sufficient on its own).[148] Providing information access to regulators – not just external auditors – increases audit quality.[149] Information access needs to be tiered to protect intellectual property and limit the risks of model leakage.[150] [151] Accessibility to civil society could increase the likelihood of innovations that meet the needs of people that are impacted by its use, for example, through understanding public perceptions of risks and perceived benefits of technologies. Foundation model regulation needs to strike a risk-benefit balance.
- Enforce a foundation model pre-market approval process, shifting the burden of proof to developers. If the regulator has the power to stop the development or sale of products, this significantly increases developers’ incentive to provide sufficient safety information. The regulatory burden needs to be distributed across the supply chain – with requirements in line with the risks at each layer of the supply chain. Cross-context risks and those with the most potential for wide-scale proliferation need to be regulated upstream at the foundation model layer; context-dependent risks should be addressed downstream in domain-specific regulation.
‘Drawing from very clear examples of real harm led the FDA to put the burden of proof on the developers – in AI this is flipped. We are very much in an ex post scenario with the burden on civil society.’
Co-Founder of Leading AI thinktank
‘We should see a foundation model as a tangible, auditable product and process that starts with the training data collection as the raw input material to the model.’
Kasia Chmielinski, Harvard Berkman Klein Center for Internet & Society
Learning through approval gates
The FDA’s capabilities have increased over time. Much of this has occurred through setting approval gates, which become points of learning for regulators. Given the novelty of foundation models and the lack of an established ‘state of the art’ for safe development and deployment, a similar approach could be taken to enhance the expertise of regulators and external evaluators (see Figure 2).
Approval gates can provide regulators with key information throughout the foundation model supply chain.
Some approval gates already exist under current sectoral regulation for specific downstream domains. At the application layer of a foundation model’s supply chain, the context of its use will be more clear than at the developer layer. Approval gates at this stage could require evidence similar to clinical studies for medical devices, to approximate risks. This could be gathered, for example, through an observational study on the automated allocation of physicians’ capacity based on described symptoms.
Current sectoral regulators may need additional resources, powers and support to appropriately evaluate the evidence and make a determination of whether a foundation model is safe to pass an approval gate.
Every time a foundation model is suggested for use, companies may already need to – or should – collect sufficient context-specific safety evidence and provide it to the regulator. For the healthcare capacity allocation example above, existing FDA – or MHRA (Medicines and Healthcare products Regulatory Agency, UK) – requirements and approval gates on clinical decision support software currently support extensive evaluation of such applications.[152]
Upstream stages of the foundation model supply chain, in particular, lack an established ‘state of the art’ defining industry standards for development and underpinning regulation. A gradual process might therefore be required to define approval requirements and the exact location of approval gates.
Initially, lighter approval requirements and stronger transparency requirements will enable learning for the regulator, allowing it to gradually set optimal risk-reducing approval requirements. The model access required by the regulator and third parties for this learning could be provided via mechanisms such as sandboxes, audits or red teaming, detailed below.
Red teaming is an approach originating in computer security. It describes exercises where individuals or groups (the ‘red team’) are tasked with looking for errors, issues or faults with a system, by taking on the role of a bad actor and ‘attacking’ it. In the case of AI, it has increasingly been adopted as an approach to look for risks of harmful outputs from AI systems.[153]
Once regulators have agreed inclusive[154] international standards and benchmarks for testing of upstream capabilities and risks, they should impose standardised thresholds for approval and endpoints. Until that point, transparency and scrutiny should be increased, and the burden of proof should be on developers to prove safety to regulators at approval gates.
The next section discusses in more specific detail how FDA-style processes could be applied to foundation model governance.
‘We need end-to-end oversight along the value chain.’
CEO of an Algorithmic auditing firm
Applying specific FDA-style processes along the foundation model supply chain
Risks can manifest across the AI supply chain. Foundation models and downstream applications can have problematic behaviours originating in pre-training data, or they can develop new ones when integrated into complex environments (like a hospital or a school). This means that new risks can emerge over time.[155] Policymakers, researchers, industry and the public therefore ‘require more visibility into the risks presented by AI systems and tools’.
Regulation can ‘play an important role in making risks more visible, and the mitigation of risk more actionable, by developing policy to enable a robust and interconnected evaluation, auditing, and disclosure ecosystem that facilitates timely accountability and remediation of potential harms’.[156]
The FDA has processes, regulatory powers and a culture that helps to identify and mitigate risks across the development and deployment process, from pre-design through to post-market monitoring. This holistic approach provides lessons for the AI regulatory ecosystem.
There are also significant similarities between specific FDA oversight mechanisms and proposals for oversight in the AI space, suggesting that the latter proposals are generally feasible. In addition, new ideas for foundation model oversight can be drawn from the FDA, such as in setting endpoints that determine the evidence required to pass an approval gate. This section draws out key lessons that AI regulators could take from the FDA approach and applies them to each layer of the supply chain.
Data and compute layers oversight
There is an information asymmetry between governments and AI developers. This is demonstrated, for example, in the way that governments have been caught off-guard by the release of ChatGPT. This also has societal implications in areas like the education sector, where universities and schools are having to respond to a potential increase in students’ use of AI-generated content for homework or assessments.[157]
To be able to anticipate these implications, regulators need much greater oversight on the early stages of foundation model development, when large training runs (the key component of the foundation model development process) and the safety precautions for such processes are being planned. This will allow greater foresight over potentially transformative AI model releases, and early risk mitigation.
Pre-submissions and Good Documentation Practice
At the start of the development process, the FDA uses pre-submissions (pre-subs), which allow it to conduct ‘risk determination’. This benefits the developer because they can get feedback from the regulator at various points, for example on protocols for clinical studies. The aim is to provide a path from device conceptualisation through to placement on the market.
This is similar to an idea that has recently gained some traction in the AI governance space: that labs should submit reports to regulators ‘before they begin the training process for new foundation models, periodically throughout the training process, and before and following model deployment’. [158]
This approach would enable learning and risk mitigation by giving access to information that currently resides only inside AI labs (and which has not so far been voluntarily disclosed), for example covering compute and capabilities evaluations,[159] what data is used to train models, or environmental impact and supply chain data.[160] It would mirror the FDA’s Quality Management System (QMS), which documents compliance with standards (ISO 13485/820) and is based on Good Documentation Practice throughout the development and deployment process to ensure risk mitigation, validation and verification, and traceability (to support regulators in the event of recall or investigations).
As well as documenting compliance in this way, the approach means that the regulator would need to demonstrate similar good practice when handling pre-submissions. Developers would have concerns around competition: the relevant authorities would need to be legally compelled to observe confidentiality, to protect intellectual property rights and trade secrets. A procedure for documenting and submitting high-value information at the compute and data input layer would be the first step towards an equivalent to the FDA approach in the AI space.
Transparency via Unique Device Identifiers (UDIs)
The FDA uses UDIs for medical devices and stand-alone software. The aim of this is to support monitoring and reporting throughout the lifecycle, particularly to identify the underlying causes of ‘adverse events’ and what corrective action should be taken (this is discussed further below).[161] This holds some similarities to AI governance proposals, particularly the suggestion for compute verification to help ensure that (pre-) training rules and safety standards are being followed.
Specifically for the AI supply chain, this would apply at the developer layer, to the essential hardware used to train and run foundation models: compute chips. Chip registration and monitoring has gained traction because, unlike other components of AI development, this hardware can be tracked in the same manner as other physical goods (like UDIs). It is also seen as an easy win. Advanced chips are usually tagged with unique numbers, so regulators would simply need to set up a registry; this could be updated each time the chips change hands.[162]
Such a registry would enable targeted interventions. For example, Jason Matheny, the CEO of RAND suggests that regulators should ‘track and license large concentrations of AI chips’, while ‘cloud providers, who own the largest clusters of AI chips, could be subject to ‘know your customer’ (KYC) requirements so that they identify clients who place huge rental orders that signal an advanced AI system is being built’.[163]
This approach would allow regulators and relevant third parties to track use throughout the lifecycle – starting with monitoring for large training runs to build advanced AI models and to verify safety compliance (for example, via KYC checks or providing information about the cybersecurity and risk management measures) for these training runs and subsequent development decisions. It would also support them to hold developers accountable if they do not comply.
Quality Management System (QMS)
The FDA’s quality system regulation is sometimes wrongly assumed to be only a ‘compliance checklist’ to be completed before the FDA approves a product. In fact, the QMS – a standardised process for documenting compliance – is intended to put ‘processes, trained personnel, and oversight’ in place to ensure that a product is ‘predictably safe throughout its development and deployment lifecycles’.
At the design phase, controls consist of design planning, design inputs that establish user needs and risk controls, design outputs, verification to ensure that the product works as planned, validation to ensure that the product works in its intended setting, and processes for transferring the software into the clinical environment.[164]
To apply a QMS to foundation model development phase, it is logical to look at the data used to (pre-)train the model. This – alongside compute – is the key input at this layer of the AI supply chain. As with the pharmaceuticals governed by the FDA, the inputs will strongly shape the outputs, such as decisions on size (of dataset and parameters), purpose (while pre-trained models are designed to be used for multiple downstream tasks, some models are better suited than others to particular types of tasks) and values (for example, choices on filtering and cleaning the data).[165]
These decisions can lead to issues in areas such as bias,[166] copyright[167] and AI-generated data[168] throughout the lifecycle. Data governance and documentation obligations are therefore needed, with similar oversight to the FDA QMS for SaMD. This will build an understanding of where risks and harms originate and make it easier to stop them from proliferating by intervening upstream.
Regulators should therefore consider model and dataset documentation methods[169] for pre-training and fine-tuning foundation models. For example, model cards document information about the model’s architecture, testing methods and intended uses,[170] while datasheets document information about a dataset, including what kind of data is included and how it was collected and processed.[171] A comprehensive model card should also contain a risk assessment,[172] similar to the FDA’s controls for testing for effectiveness in intended settings. This could be based on uses foreseen by foundation model developers. Compelling this level of documentation would help to introduce FDA-style levels of QMS practice for AI training data.
Core policy implications
An approach to pre-notification of, and information-sharing on, large training runs could use the pre-registration process of the FDA as a model. As discussed above, under the FDA regime, developers are continuously providing information to the regulator, from the pre-training stage onwards.[173] This should also be the case in relation to foundation models.
It might also make sense to track core inputs to training runs by giving UDIs to microchips. This would allow compliance with regulations or standards to be tracked and would ensure that the regulator would have sight of non-notified large training runs. Finally, the other key input into training AI models – data – should adhere to documentation obligations, similarly to FDA QMS procedures.
Foundation model developer layer oversight
Decisions taken early in the development process have significant implications downstream. For example, models (pre-)trained on fundamental human rights values produce outputs that are less structurally harmful.[174] To reduce risk of harm as early as possible, critical decisions that shape performance across the supply chain should be documented as they are made, before wide-scale distribution, fine-tuning or application,
Third-party evidence generation and endpoints
The FDA model relies on third-party efficacy and safety evidence to prove ‘endpoints’ (targeted outcomes, jointly agreed between the FDA and developers before a clinical trial) as defined in standards or in an exploratory manner together with the FDA. This allows high-quality information on the pre-market processes for devices to be gathered and submitted to regulators.
Narrowly defined endpoints are very similar to one of the most commonly cited interventions in the AI governance space: technical audits.[175] A technical audit is ‘a narrowly targeted test of a particular hypothesis about a system, usually by looking at its inputs and outputs – for instance, seeing if the system performs differently for different user groups’. Such audits have been suggested by many AI developers and researchers and by civil society.[176]
Regulators should therefore develop – or support the AI ecosystem to develop – benchmarks and metrics to assess the capabilities of foundation models, and possibly thresholds that a model would have to meet before it could be placed on the market. This would help standardise the approach to third-party compliance with evidence and measurement requirements, as under the FDA, and establish a culture of safety in the sector.
Clinical trials
In the absence of narrowly defined endpoints and in cases of uncertainty, the FDA works with developers and third-party experts to enable more exploratory scrutiny as part of trials and approvals. Some of these trials are based on iterative risk management and explorative auditing, and on small-scale deployment to facilitate ‘learning by doing’ on safety issues. This informs what monitoring is needed, provides iterative advice and leads to learning being embedded in regulations afterwards.
AI regulators could use similar mechanisms, such as (regulatory) sandboxes. This would involve pre-market, small-scale deployment of AI models in real-world but controlled conditions, with regulator oversight.
This could be done using a representative population for red-teaming, expert ‘adversarial’ red-teamers (at the foundation model developer stage), or sandboxing more focused on foreseeable or experimental applications and how they interact with end users. In some jurisdictions, existing regulatory obligations could be used as the endpoint and offer presumptions of conformity – and therefore market access – after sandbox testing (as in the EU AI Act).
It will take work to develop a method and an ecosystem of independent experts who can work on third-party audits and sandboxes for foundation models. But this is a challenge the FDA has met, as have other sectors such as aviation, motor vehicles and banking.[177] An approach like the one described above has been used in aviation to monitor and document incidents and devise risk mitigation strategies. This helped to encourage a culture of safety in the industry, reducing fatality risk by 83 per cent between 1998 and 2008 (at the same time as a five per cent annual increase in passenger kilometres flown).[178]
Many organisations already exist that can service this need in the AI space (for example, Eticas AI, AppliedAI, Algorithmic Audit, Apollo Research), and more are likely to be set up.[179]
An alternative to sandboxes is to consider structured access for foundation models, at least until it can be proven that a model is safe for wide-scale deployment.[180] This would be an adaptation of the FDA’s approach to clinical trials, which allows experimentation with a limited number of people when the technology has a wide spectrum of uses (for example, gene editing) or when the risks are unclear, to get insights while preventing any harms that arise from proliferation.
Applied to AI, this could entail a staged release process – something leading AI researchers have already advocated for. This would involve model release to a small number of people (for example, vetted researchers) so that ‘beta’ testing is not done on the whole population via mass deployment.
Internal testing and disclosure of ‘adverse events’
Another mechanism used at the development stage by the FDA is internal testing and mandatory disclosure of ‘adverse events’. Regulators could impose similar obligations on foundation model developers, requiring internal audits and red teaming[181] and the disclosure of findings to regulators. Again, these approaches have been suggested by leading AI developers.[182] They could be made more rigorous by coupling them with mandatory disclosure, as under the FDA regime.
The AI governance equivalent of reporting ‘adverse effects’ might be incident monitoring.[183] This would involve a ‘systematic approach to the collection and dissemination of incident analysis to illuminate patterns in harms caused by AI’.[184] The approach could be strengthened further by including ‘near-miss’ incidents.[185]
In developing these proposals, however, it is important to bear in mind challenges faced in the life sciences sector regarding how to make adverse effect reporting suitably prescriptive. For example, clear indicators for what to report need to be established so that developers cannot claim ignorance and underreport.
However, it is not possible to foresee all potential effects of a foundation model. As a result, there needs to be some flexibility in incident reporting as well as penalties for not reporting. Medical device regulators in the UK have navigated this by providing high-level examples of indirect harms to look out for, and examples of the causes of these harms.[186] In the USA, drug and device developers are liable to report larger-scale incidents, enforced by the FDA through, for example, fines. If enacted effectively, this kind of incident reporting would be a valuable foresight mechanism for identifying emergent harms.
A pre-market approval gate for foundation models
After the foundation model developer layer, regulators should consider a pre-market approval gate (as used by the FDA) at the point just before the model is made widely available and accessible for use by other businesses and consumers. This would build on the mandatory disclosure obligations at the data and compute layers and involve submitting all documentation compiled from third-party audits, internal audits, red teaming and sandbox testing. It would be a rigorous regime, similar to the FDA’s use of QMS, third-party efficacy evidence, adverse event reporting and clinical trials.
AI regulators should ensure that documentation and testing practices are standardised, as they are in FDA oversight. This would ensure that high-value information is used for market approval at the optimal time, to minimise the risk of potential downstream harms before a model is released onto the market.
This approach also depends on developing adequate benchmarks and standards. As a stopgap, approval gates could initially be based on transparency requirements and the provision of exploratory evidence. As benchmarks and standards emerged over time, the evidence required could be more clearly defined.
Such an approval gate would be consistent with one of the key risk-reducing features of the FDA’s approach: putting the burden of proof on developers. Many of the concerns around third-party audits of foundation models (in the context of the EU AI Act) centre on the lack of technological expertise beyond AI labs. A pre-market approval gate would allow AI regulators to specify what levels of safety they expect before a foundation model can reach the market, but the responsibility for proving safety and reliability would be placed on the experts who wish to bring the model to market.
In addition, the approval gate offers the regulator and accredited third parties the chance to learn. As the regulator learns – and the technology develops – approval gates could be updated via binding guidance (rather than legislative changes). This combination of ‘intervention and reflection’ has ‘been shown to work in safety-critical domains such as health’.[187] Regulators and other third parties should cascade this learning downstream, for example, to parties who build on top of the foundation model. This is a key risk-reducing feature of the FDA’s approach: the ‘approvers’ and others in the ecosystem become more capable and more aware of safe use and risk mitigation.
While the burden of proof would be primarily on developers (who may use third parties to support in evidence creation), approval would still depend on the regulator. Another key lesson from FDA processes is that the regulator should bring in support from independent experts in cases of uncertainty, via a committee of experts, consumer and industry representatives, and patient representatives. This is important, as the EU’s regulatory regime for AI has been criticised for a lack of multi-stakeholder governance mechanisms, including ‘effective citizen engagement’.[188]
Indeed, many commercial AI labs say that they want avenues for democratic oversight and public participation (for example, OpenAI and Anthropic’s participation in ‘alignment assemblies’,[189] which seek public opinion to inform, for example, release criteria) but are unclear on how to establish them.[190] Introducing ways to engage stakeholders in cases of uncertainty as part of the foundation model approval process could help to address this. It would give a voice to those who could be affected by models with potentially societal-level implications, in the same way patients are given a voice in FDA review processes for SaMD. It might also help address one of the limitations of the FDA: an overreliance on industry expertise in some novel areas.
To introduce public participation in foundation model oversight in a meaningful way, it would be important to consider the approach to engagement that is suitable to help to identify risks.
One criteria to consider is who should be involved, with options ranging from a representative panel or jury of members of the public to panels formed of members of the public at higher risk of harm or marginalisation.
Another criteria to consider relates to the depth of engagement. The depth of engagement is often framed as a spectrum from low involvement, such as public consultations, all the way to deeper processes that involve partnership in decision-making.[191]
A third criteria to consider is the method of engagement. This would depend on decisions related to who should be involved and to what extent. For example, surveys or focus groups are common in consultative exercises, workshops can enable more involvement whereas panels and juries allow for deeper engagement which can result in its members proposing recommendations. In any case it will be important to consider whose voices, experiences and potential harms will be included or missed, and ensure those less represented or at more risk of harms are part of the process.
Finally, there are ongoing debates about whether pre-market approval should be applied to all foundation models, or ‘tiered’ to ensure those with the most potential to impact society are subject to greater oversight.
While answering this question is beyond the scope of this paper, it seems important that both ex ante and ex post metrics are considered when establishing which models belong in which tier. The former might include, for example, measurement of modalities, the generality of the base model, the distribution method and the potential for adaptation of the model. The latter could include the number of downstream applications built on the model, the number of users across applications and how many times the model is being queried. Any regulator must have the power and capacity to update the makeup of tiers in a timely fashion as and when these metrics shift.
Application layer oversight
Following the AI supply chain, a foundation model is made available and distributed via the ‘host’ layer, by either the model provider (API access) or a cloud service provider (for example, Hugging Face, which hosts models for download).
Some argue that this layer should also have some responsibility for the safe development and distribution of foundation models (for example, through KYC checks, safety testing before hosting or take-down obligations in case of harm). But there is a reason why regulators have focused primarily on developers and deployers: they have the most control over decisions affecting risk origin and safety levels. For this reason, we also focus on interventions beyond the host layer.
However, a minimal set of obligations on host layer actors (such as cloud service providers or model hosting platforms) is necessary, as they could play a role in evaluating model usage, implementing trust and safety policies to remove models that have demonstrated or are likely to demonstrate serious risks, and flagging harmful models to regulators when it is not in their power to take them down. This is beyond the scope of this paper, and we suggest that the responsibilities of the host layer are addressed in further research.
Once a foundation model is on the market and it is fine-tuned, built upon or deployed by downstream users, its risk profile becomes clearer. Regulatory gates and product safety checks are introduced by existing regulators at this stage, for example in healthcare, automotives or machinery (see UK regulation of large language models – LLMs – as medical devices, or the EU AI Act’s regulation of foundation models deployed in ‘high-risk’ areas). These are useful regulatory endpoints that should help to reduce risk and harm proliferation.
However, there are still lessons to be learned at the application layer from the FDA model. Many of the mechanisms used at the foundation model developer layer could be used at this layer, but with endpoints defined based on the risk profile of the area of deployment. This could take the form of third-party audits based on context-specific standards, or sandboxes including representative users based on the specific setting in which the AI system will be used.
Commercial off-the-shelf software (COTS) in critical environments
One essential mechanism for the application layer is a deployment risk assessment. Researchers have proposed that this should involve a review of ‘(a) whether or not the model is safe to deploy, and (b) the appropriate guardrails for ensuring the deployment is safe’.[192] This would serve as an additional gate for context-specific risks and is similar to the FDA’s rules for systems that integrate COTS in severe-risk environments. Under these rules, additional approval is needed unless the COTS is approved for use in that context.
A comparable AI governance regime could allow foundation models that pass the earlier approval gate to be used downstream unless they are to be used in a high-risk or critical sector, in which case a new risk assessment would have to be undertaken and further regulatory approval sought.
For example, foundation models applied in critical energy system would be pre-approved as COTS. The final approval would still need to be given by energy regulators, but the process would be substantially easier for pre-approved COTS. The EU AI Act employs a similar approach: foundation models that are given a high-risk ‘intended purpose’ by downstream developers would have to undergo EU conformity assessment procedures.
Algorithmic impact assessments are a tool for assessing the possible societal impacts of an AI system before the system is in use (with ongoing monitoring often advised).[193] Such assessments should be undertaken when an AI system is to be deployed in a critical area such as cybersecurity, and mitigation measures put in place. This assessment should be coupled with a new risk assessment (in addition to that carried out by the foundation model developer), tailored to the area of deployment. This could involve additional context-specific guidance or questions from regulators, and the subsequent mitigation measures should address these.
Algorithmic impact and risk assessments are essential components at the application layer for high-risk deployments, and are very similar to the QMS imposed by the FDA throughout the development and deployment process. If they are done correctly, they can help to ensure that risk and impact mitigation measures are put in place to cover the lifecycle and will form the basis of post-market monitoring processes.
Some AI governance experts have suggested that these assessments should be complemented by user evaluation and testing – defined as assessments of user-centric effects of an application or system, its functionality and its restrictions, usually via user testing or surveys.[194] These evaluations could be tailored to the intended use context of an application, to ensure adequate representation of people potentially affected by it, and would be similar to the context-specific audit gates used by the FDA.
Post-market monitoring
Across sectors, one-off conformity checks have been shown to open the door for regulations to be ‘gamed’ or for emergent behaviours to be missed (see the Volkswagen emissions scandal).[195] These issues are even more likely to arise in relation to AI, given its dynamic nature, including the capacity to change throughout the lifecycle and for downstream users to fine-tune and (re)deploy models in complex environments. The FDA model shows how these risks can be reduced by having an ecosystem of reporting and foresight, and strong regulatory powers to act to mitigate risks.
MedWatch and MedSun reporting
Post-market monitoring by the FDA includes reporting mechanisms such as MedWatch and MedSun.[196] These mechanisms enable adverse event reporting for medical products, as well as monitoring of the safety and effectiveness of medical devices. Serious incidents are documented and their details made available to consumers.
In the AI space, there are similar proposals for foundation model developers, and for high-risk application providers building on top of these models, to implement ‘an easy complaint mechanism for users and to swiftly report any serious risks that have been identified’.[197] This should compel the upstream providers to take corrective action when they can, and to document and report serious incidents to regulators.
This is particularly important for foundation models that are provided via API, as in this case the provider maintains a huge degree of control over the underlying model.[198] This would mean that the provider would usually be able to mitigate or correct the emerging risk. It would also reduce the burden on regulators to document incidents or take corrective action. Leading AI developers have already committed to introducing a ‘robust reporting mechanism’ to allow ‘issues [that] may persist even after an AI system is released’ to be ‘found and fixed quickly’.[199] Regulators could consider putting such a regime in place for all foundation models.
Regulators could also consider detection mechanisms for generative foundation models. These would aim to ‘distinguish content produced by the foundation model from other content, with a high degree of reliability’, as recently proposed by the Global Partnership on AI.[200] Their report found that this is ‘technically feasible and would play an important role in reducing certain risks from foundation models in many domains’. Requiring this approach, at least for the largest model providers (who have the resources and expertise to develop detection mechanisms), could mitigate risks such as disinformation and subsequent undermining of the rule of law or democracy.
Other reporting mechanisms for foundation models have been proposed, which overlap with the FDA’s ‘usability and clinical data logging, and trend reporting’. For example, Stanford researchers have suggested that regulators should compel the disclosure of usage patterns, in the same manner of transparency reporting for online platforms.[201] This would greatly enhance understanding of ‘how foundation models are used (for example, for providing medical advice, preparing legal documents) to hold their providers to account’.[202]
Concern-based audits
Concern-based audits are a key part of the FDA’s post-market governance. They are triggered by real-world monitoring of consumers and impacts after approval. If concerns are identified, the FDA has strong enforcement mechanisms that allow it to access relevant data and documentation. The audits are rigorous and have been shown to have strong deterrence effects on negligent behaviour by drug companies.
Mechanisms for highlighting ‘concern’ in the AI space could include reporting mechanisms and ‘trusted flaggers’ – organisations that are formally recognised as independent, and with the requisite expertise, for identifying and reporting concerns. People affected by the technologies could be given the right to lodge a complaint with supervisory authorities, such as an AI ombudsman, to support people affected by AI and increase regulators’ awareness of AI harms as they occur.[203] [204] . This should be complimented by a comprehensive remedies framework for affected persons based on effective avenues for redress, including a right to lodge a complaint with a supervisory authority, judicial remedy and an explanation of individual decision-making
Feedback loops
Post-market monitoring is a critical element of the FDA’s risk-reducing features. It is based on mechanisms to facilitate feedback loops between developers, regulators, practitioners and patients. As discussed above, Unique Device Identifiers at the pre-registration stage support monitoring and traceability throughout the lifecycle, while ongoing review of quality, safety and efficacy data via QMS further supports this. Post-market monitoring for foundation models should similarly facilitate such feedback loops. These could include customer feedback, usability and user prompt screening, human-AI interaction evaluations and cross-company reporting of trends and structural indicators. Beyond feedback to the provider, affected persons should also be able to report incidents directly to a regulatory authority, particularly where harm arises, or is reasonably foreseeable to arise.
Software of Unknown Provenance (SOUP)
In the context of safety-critical medical software, SOUP is software that has been developed with an unknown development process or methodology, or which has unknown safety-related properties. The FDA monitors for SOUP by compelling the documentation of pre-specified post-market software adaptations, meaning that the regulator can validate changes to a product’s performance and monitor for issues and unforeseen use in software.[205]
Requiring similar documentation and disclosure of software and cybersecurity issues after deployment of a foundation model would be a minimum sensible safeguard for both risk mitigation and regulator learning. This could also include sharing issues back upstream to the model developer so that they can take corrective action or update testing and risk profiles.
The approach should be implemented alongside the obligations around internal testing and disclosure of adverse events for foundation models at the developer layer. Some have argued that disclosure of near misses should also be required (as it is in the aviation industry)[206] as an added incentive for safe development and deployment.
Another parallel with the monitoring of SOUP can be seen in AI governance proposals for measures around open-source foundation models. To reduce the unknown element, and for transparency and accountability reasons, application providers – or whoever makes the model or system available on the market – could be required to make it clear to affected persons when they are engaging with AI systems and what the underlying model is (including if it is open source), and to share easily accessible explanations of systems’ main parameters and any opt-out mechanisms or human alternatives available.[207] This would be the first step to both corrective action to mitigate risk or harm, and redress if a person is harmed. It is also a means to identify the use of untested underlying foundation models.
Finally, similar to the FDA’s use of documentation of pre-specified, post-market software adaptations, AI regulators could consider mandating that developers and application deployers document and share planned and foreseeable changes downstream. This would have to be defined clearly and standardised by regulators to a proportionate level, taking into consideration intellectual property and trade secret concerns, and the risk of the system being ‘gamed’ in the context of new capabilities. In other sectors, such as aviation, there have been examples of changes being underreported to avoid new costs, such as retraining.[208] But a similar regime would be particularly relevant for AI models and systems, given their unique ability to learn and develop throughout their lifecycle.
The need for documenting or pre-specifying post-market adaptations of foundation models could be based on capabilities evaluations and risk assessments, so that new capabilities or risks that arise post-deployment are reported to the ecosystem. Significant changes could trigger additional safety checks, such as third-party (‘concern-based’, in FDA parlance) audits or red teaming to stress-test the new capabilities.
Investigative powers
The FDA’s post-market monitoring puts reporting obligations on providers and users, while underpinning this with strong investigative powers. It conducts ‘active surveillance’ (for example, under the Sentinel Initiative),[209] and it is legally empowered to check QMS and other documentation and logging data, request comprehensive evidence and conduct inspections.
Similarly, AI regulators should have powers to investigate foundation model developers and downstream deployers, such as for monitoring and learning purposes or when investigating suspected non-compliance. This could include off- and on-site inspections to gather evidence, to address the information asymmetries between AI developers and regulators, and to mitigate emergent risks or harms.
Such a regime would require adequate resources and sociotechnical expertise. Foundation models are a general-purpose technology that will increasingly form part of our digital infrastructure. In this light, there needs to be a recognition that regulators should be funded on a comparable level to other domains in which safety and public trust are paramount and where underlying technologies form important parts of national infrastructure – such as civil nuclear, civil aviation, medicines, and road and rail.[210]
Recalls, market withdrawals and safety alerts
The FDA uses recalls, market withdrawals and safety alerts when products are in violation of law. Recall can also be a voluntary action by manufacturers and distributors to meet their responsibility to protect public health and wellbeing from products that present risk or are otherwise defective.[211]
Some AI governance experts and standards bodies have called for foundation model developers to similarly establish standard criteria and protocols for when and how to restrict, suspend or retire a model from active use.[212] This would be based on monitoring by the original providers throughout the lifecycle for harmful impacts, misuse or security vulnerabilities (including leaks or otherwise unauthorised access).
Whistleblower protection
In the same way that the FDA mandates reporting, with associated whistleblower protections, of adverse events by employees, second-party clinical trial conductors and healthcare practitioners, AI regulators should protect whistleblowers (for example, academics, designers, developers, project contributors, auditors, product managers, engineers and economic operators) who suspect breaches of law by a developer or deployer or an AI model or system. This protection should be developed in a way that learns from the pitfalls of whistleblower law in other sectors, which have led to ineffective uptake or enforcement. This includes ensuring breadth of coverage, clear communication of processes and protections, and review mechanisms.[213]
Recommendations and open questions
The FDA model of pre-approval and monitoring is an important inspiration for regulating novel technologies with potentially severe risks, such as foundation models.
This model entails risk-based mandates for pre-approval based on mandatory safety evidence. This works well when risks reliably originate and can be identified before proliferating or developing into harms.
The general-purpose nature of foundation models requires exploratory external scrutiny upstream in the supply chain, and targeted sector-specific approvals downstream.
Risks need to be identified and mitigated before they proliferate. This is especially difficult for foundation models.[214] Explorative approval gates have been ‘shown to work in safety-critical domains such as health’, due to the combination of ‘intervention and reflection’. Pre-approvals offer the FDA a mechanism for intervention, allowing most risks to be caught.
Another important feature of oversight is reflection. In health regulation, this is achieved through ‘iteration via guidance, rather than requiring legislative changes’.[215] This is a key consideration for AI regulators, who should be empowered (and compelled) to frequently update rules via binding guidance.
A continuous learning process to build suitable approval and monitoring regimes for foundation models is essential, especially at the model development layer. Downstream, there needs to be targeted scrutiny and approval for deployment through existing approval gates in specific application areas.
Effective oversight of foundation models requires recurring, independent evaluations and audits and access to information, placing the burden of proof on developers – not on civil society or regulators.
Literature reviews of other industries[216] show that this might be achieved through risk-based reviews by empowered regulators and third parties, tiered access for evaluators, mandatory pre-approvals, and treating foundation models like auditable products.
Our general principles for AI regulators are detailed in the section ‘Applying key features of FDA-style oversight to foundation models’.
Recommendations for AI regulators, developers and deployers
Data and compute layers oversight
- Regulators should compel pre-notification of, and information-sharing on, large training runs. Providers of compute for such training runs should cooperate with regulators on monitoring (by registering device IDs for microchips) and safety verification (KYC checks and tracking).
- FDA inspiration: pre-submissions, Unique Device Identifiers (UDIs)
- Regulators should compel mandatory model and dataset documentation and disclosure for the pre-training and fine-tuning of foundation models,[217] [218] [219] including a capabilities evaluation and risk assessment within the model card for the (pre-) training stage and throughout the lifecycle.[220] Dataset documentation should focus on a description of training data that is safe to be made public (what is in it, where was it collected, under what licence, etc.), coupled with structured access for regulators or researchers to the training data itself (while adhering to strict levels of cybersecurity, as even this access carries security risks).
- FDA inspiration: Quality Management System (QMS)
Foundation model layer oversight
- Regulators should introduce a pre-market approval gate for foundation models, as this is the most obvious point at which risks can proliferate. In any jurisdiction, defining the approval gate will require significant work, with input from all relevant stakeholders. Clarity should be provided about which foundation models would be subject to this stricter form of pre-market approval. Based on the FDA findings, this gate should at least entail submission of evidence to prove safety and market readiness based on internal testing and audits, third-party audits and (optional) sandboxes. Making models available on a strict and controllable basis via structured access could be considered as a temporary fix until an auditing ecosystem and/or sandboxes are developed.Depending on the jurisdiction in question and existing or foreseen pre-market approval for high-risk use, an additional approval gate should be introduced using endpoints (outcomes or thresholds to be met to determine efficacy and safety) based on the risk profile of the area of deployment for the application layer.
- FDA inspiration: QMS, third-party efficacy evidence, adverse events reporting, clinical trials
- Third-party audits should be required as part of the pre-market approval process, and sandbox testing (as described in Recommendation 3) in real-world conditions should be considered. These should consist of – at least – a third-party audit based on context-specific standards. Alternatively, regulators could use sandboxes that include representative users (based on the setting in which the AI system will be used) to check conformity before deployment. Results should be documented and disclosed to the regulator.
- FDA inspiration: third-party efficacy evidence, adverse events reporting, clinical trials
- Developers should enable detection mechanisms for outputs of generative foundation models.[221] Developers and deployers should make clear to affected persons and end users when they are engaging with AI systems. As an additional safety mechanism, they should build in detection mechanisms to allow end users and affected persons to ‘distinguish content produced by the foundation model from other content, with a high degree of reliability’.[222] Such detection mechanisms are important both as a defensive tool (for example, tagging AI-generated content) and also to enable study of model impacts. AI regulators could consider making this mandatory, at least for the most significant models (developers of which may have the resources and expertise to develop detection mechanisms).
- FDA inspiration: post-market safety monitoring
- As part of the initial risk assessment, developers and deployers should document and share planned and foreseeable modifications throughout the foundation model’s supply chain. A substantial modification that falls outside this scope should trigger additional safety checks, such as third-party (‘concern-based’) audits or red teaming to stress test the new capabilities.
- FDA: concern-based audits, pre-specified change control plans
- Foundation model developers, and subsequently high-risk application providers building on top of these models, should enable an easy complaint mechanism for users to swiftly report any serious risks that have been identified. This should compel upstream providers to take corrective action when they can, and to document and report serious incidents to regulators. These feedback loops should be strengthened further by awareness-raising across the ecosystem about reporting, and sharing lessons learned on what has been reported and corrective actions taken.
- FDA Inspiration: MedWatch and MedSun programs
Application layer oversight
- Existing sector-specific agencies should review and approve the use of foundation models for a set of use cases, by risk level. Deployers of foundation models in high-risk or critical areas (to be defined in each jurisdiction) should undertake a deployment risk assessment to review ‘(a) whether or not the model is safe to deploy, and (b) the appropriate guardrails for ensuring the deployment is safe’.[223] Upstream developers should cooperate and share information with downstream customers to conduct this assessment. If the model is deemed safe, they should also undertake an algorithmic impact assessment to assess possible societal impacts of an AI system before the system is in use (with ongoing monitoring often advised).[224] Results should be documented and disclosed to the regulator.
- FDA inspiration: COTS (commercial off-the-shelf software), QMS
- Downstream application providers should make clear to end users and affected persons what the underlying foundation model is, including if it is an open-source model, and provide easily accessible explanations of systems’ main parameters and any opt-out mechanisms or human alternatives available.[225]
- FDA inspiration: Software of Unknown Provenance (SOUP)
Post-market monitoring
- An AI ombudsman should be considered, to receive and document complaints or known instances of harms of AI. This would increase regulators’ visibility of AI harms as they occur. It could be piloted initially for a relatively modest investment, but if successful it could dramatically improve redress for AI harms and the functionality of an AI regulatory framework as a whole.[226] An ombudsman should be complimented by a comprehensive remedies framework for affected persons based on clear avenues for redress.
- FDA inspiration: concern-based audits, reporting of adverse events
- Developers and deployers should provide documentation and disclosure of incidents throughout the supply chain, including near misses.[227] This could be strengthened by requiring downstream developers (building on top of foundation models at the application layer) and end users (for example, medical or education professionals) to also disclose incidents.
- FDA inspiration: reporting of adverse events
- Foundation model developers and downstream deployers should be compelled to restrict, suspend or retire a model from active use if harmful impacts, misuse or security vulnerabilities (including leaks or other unauthorised access) arise. Such decisions should be based on standardised criteria and processes.[228]
- Host layer actors (for example, cloud service providers or model hosting platforms) should also play a role by evaluating model usage, implementing trust and safety policies to remove models that have demonstrated or are likely to demonstrate serious risks, and flagging harmful models to regulators when it is not in their power to take them down.
- FDA inspiration: recalls, market withdrawals and safety alerts
- AI regulators should have strong powers to investigate and require evidence generation from foundation model developers and downstream deployers. This should be strengthened by whistleblower protections for anyone involved in the development or deployment process who raises concerns about risks to health or safety. This would support regulatory learning and act as a strong deterrent to rule breaking. Powers should include off- and on-site inspections and evidence-gathering mechanisms to address the information asymmetries between AI developers and regulators and to mitigate emergent risks or harms. Consideration should be given to the trade-offs between intellectual property, trade secret and privacy protections (and whether these could serve as undue legal loopholes) and the safety-enhancing features of investigative powers: regulators considering the FDA model across jurisdictions should clarify such legally contentious issues.
- FDA inspiration: wide information access, active surveillance
- Any regulator should be funded to a level comparable to (if not greater than) regulators in other domains where safety and public trust are paramount and where underlying technologies form part of national infrastructure – such as civil nuclear, civil aviation, medicines, or road and rail.[229] Given the level of resourcing required, this may be partly funded by AI developers over a certain threshold (to be defined the regulatorfor example, annual turnover)– as is the case with the FDA[230] and the EU’s European Medicines Agency (EMA).[231] Such an approach is important, to ensure that regulators have a source of funding that is stable and secure, and (importantly) independent from political decisions or reprioritisation.
- FDA inspiration: mandatory fees
- The law around AI liability should be clarified to ensure that legal and financial liability for AI risk is distributed proportionately along foundation model supply chains. Liability regimes vary between jurisdictions and a thorough assessment is beyond the scope of this paper, but across sectors regulating complex technology, clarity in liability is a key driver of compliance within companies and uptake of the technology. For example, lack of clarity as to end user liability in clinical AI is a major reason that uptake has been limited. Liability will be even more contentious in the foundation model supply chain when applications are developed on top of foundation models, and this must be addressed accordingly in any regulatory regime for AI.
Overcoming the limitations of the FDA in a prospective AI regulatory regime
Having considered how the risk-reducing mechanisms of the FDA might be applied to AI governance, it makes sense to also acknowledge the limitations of the FDA regime, and to consider how they might also be counterbalanced in a prospective AI regulatory regime.
The first limitation is the lack of coverage for systemic risks, as the FDA focuses on risk to life. Systemic risks are prevalent in the AI space.[232] AI researchers have conceptualised systemic risk as societal harm and point out that it is similarly overlooked. Proposals to address this include: ‘(1) public oversight mechanisms to increase accountability, including mandatory impact assessments with the opportunity to provide societal feedback; (2) public monitoring mechanisms to ensure independent information gathering and dissemination about AI’s societal impact; and (3) the introduction of procedural rights with a societal dimension, including a right to access to information, access to justice, and participation in public decision-making on AI, regardless of the demonstration of individual harm’.[233] We have expanded on and included these mechanisms in our recommendations in the hope that they can overcome limitations centring on systemic risks.
The second limitation is the high cost of compliance and subsequent limited number of developers, given that the stringent approval requirements are challenging for smaller players to meet. Inspiration for how to counterbalance this may be gleaned from the EU’s FDA equivalent, the EMA. It offers tailored support to small and medium-sized enterprises (SMEs), via an SME Office that provides regulatory assistance for reduced fees. This has contributed to the approval rates for SME applicants increasing from 40 per cent in 2016 to 89 per cent in 2020.[234] Similarly, the UK’s NHS has an AI & Digital Regulations Service that gives guidance and advice on navigating regulation, especially for SMEs that do not have compliance teams.[235]
Streamlined regulatory pathways could be considered to further reduce burdens for AI models or systems with demonstrably promising potential (for example, for scientific discovery). The EMA has done this through its Advanced Therapy Medicine Products process, which streamlines approval procedures for certain medicines.[236]
Similar support mechanisms could be considered for SMEs and startups, as well as streamlined procedures for demonstrably beneficial AI technology, under an AI regulator.
The third limitation is the FDA’s overreliance on industry in some novel areas, because of a lack of expertise. Lack of capacity for effective regulatory oversight has been voiced as a concern in the AI space, too.[237] Some ideas exist for how to overcome this, such as the Singaporean AI Office’s use of public–private partnerships to utilise industry talent without being reliant on it.[238]
The EMA has grappled with similar challenges. Like the FDA, it overcomes knowledge gaps by having a pool of scientific experts, but it seeks to prevent conflict of interest by leaning substantially on transparency: the EMA Management Board and experts cannot have any financial or other interests in the industry they are overseeing, and the curricula vitae, declarations of interest and risk levels for these experts are publicly available.[239]
Taken together, these solutions might be considered to reduce the chances of the limitations of FDA governance being reproduced by an AI regulator.
Open questions
The proposed FDA-style oversight approach for foundation models is far from a detailed ready-to-implement guideline for regulators. We acknowledge the small sample of interviewees for this paper, and that many of our interview subjects may strongly support an FDA model for regulation. For further validation and detailing of the claims in this paper, we are especially interested in future work on three sets of questions.
Understanding foundation model risks
- Across the foundation model supply chain, where exactly do foundation model risks[240] originate and proliferate, and which players need to be tasked with their mitigation? How can unknown risks be discovered?
- How effective will exploratory and targeted scrutiny be in identifying different kinds of risks for foundation models?
- Do current and future foundation models need to be categorised along risk tiers? If so, how? Do all foundation models need to go through an equally rigorous process of regulatory approvals?
Detailing FDA-style oversight for foundation models to foster ‘safe innovation’
- For the FDA, what aspects of regulatory guidance were easier to prescribe, and to enforce in practice?
- How do FDA-style oversight or specific oversight features address each risk of foundation models in detail?
- How can FDA-style oversight for foundation models be integrated into international oversight regimes?[241]
- What do FDA-style review, audit and inspection processes look like, step by step, for foundation models?
- How can the limitations of the FDA approach be addressed in every layer of the foundation model supply chain? How can difficult-to-detect systemic risks be mitigated? How can the stifling of innovation, especially among SMEs, be avoided?
- Are FDA-style product recalls feasible for a foundation model or a downstream applications of foundation models?
- What role should third parties in the host layer play? While they have less remit over risk origin, might they have significant control over, for example, risk mitigation?
- What are the implications of FDA-style oversight for foundation models on their accessibility, affordability and sharing their benefits?
- How would FDA-style pre-approvals be enforced for foundation models, for example, for product recalls?
- How is liability distributed in an FDA-style oversight approach?
- Why is the FDA able to be stringent/cautious? How do political incentives on congressional oversight and aversion to risk of harms of medication apply to foundation model regulation?
- What can be learned from the political economy of the FDA and its reputation?
- In each jurisdiction (for example, USA, UK, EU), how does an FDA-style approach for AI fit into the political economy and institutional landscape?
- In each jurisdiction, how should liability law be adapted for AI to ensure that legal and financial liability for AI risk is distributed proportionately along foundation model supply chains?
Learnings from other regulators
- What can be learned from regulators in public health in other jurisdictions, like the UK’s Medicines and Healthcare products Regulatory Agency (MHRA), EU’s EMA and Health Canada? [242] [243] [244]
- How can other non-health regulators, such as the US Federal Aviation Administration or National Highway Traffic Safety Administration, inspire foundation model oversight?[245]
- How can novel forms of oversight and audits, such as cross-audits or joint audits, be coupled with processes from existing regulators?
Acknowledgements
This paper was co-authored by Merlin Stein (PhD candidate at the University of Oxford) and Connor Dunlop (EU Public Policy Lead at the Ada Lovelace Institute) with input from Andrew Strait.
Interviewees
The 20 interviewees included experts on FDA oversight and foundation model evaluation processes from industry, academia, and thinktanks, as well as government officials. This included three interviews with leading AI labs, two with third-party AI evaluators and auditors, nine with civil society organisations, and six with medical software regulation experts, including former FDA leadership and clinical trial leaders.
The following participants gave us permission to mention their names and affiliations (in alphabetical order). Ten interviewees not listed here did not provide their permission. Respondents do not represent any organisations they are affiliated with. They chose to add their name after the interview and were not sent a draft of this paper before publication. The views expressed in this paper are of the Ada Lovelace Institute.
- Kasia Chmielinski, Berkman Klein Center for Internet & Society
- Gemma Galdón-Clavell, Eticas Research & Consulting
- Gilian Hadfield, University of Toronto, Vector Institute and OpenAI, independent contractor
- Sonia Khatri, independent SaMD and medical device regulation expert
- Igor Krawczuk, Lausanne Institute of Technology
- Sarah Myers West, AI Now Institute
- Noah Strait, Scientific and Medical Affairs Consulting
- Robert Trager, Blavatnik School of Government, University of Oxford, and Centre for the Governance of AI
- Alexandra Tsalidas, Harvard Ethical Intelligence Lab
- Rudolf Wagner, independent senior executive advisor for SaMD
Reviewers
We are grateful for helpful comments and discussions on this work from:
- Ashwin Acharya
- Markus Anderljung
- Clíodhna Ní Ghuidhir
- Xiaoxuan Liu
- Deborah Raji
- Sarah Myers West
- Moritz von Knebel
Footnotes
[1] ‘Voluntary AI Commitments’, <www.whitehouse.gov/wp-content/uploads/2023/09/Voluntary-AI-Commitments-September-2023.pdf>, accessed October 12, 2023
[2] ‘An EU AI Act that works for people and society’ (Ada Lovelace Institute 2023) <www.adalovelaceinstitute.org/policy-briefing/eu-ai-act-trilogues/> accessed 12 October 2023
[3] The factors that determine AI risk are not purely technical – sociotechnical determinants of risk are crucial. Features such as the context of deployment, the competency of the intended users, and the optionality of interacting with an AI system must all be considered, in addition to specifics of the data and AI model deployed. OECD, “OECD Framework for the Classification of AI Systems,” OECD Digital Economy Papers, no. 323 (February 2022), https://doi.org/10.1787/cb6d9eca-en.
[4] Markus Anderljung and others, ‘Frontier AI Regulation: Managing Emerging Risks to Public Safety’ (arXiv, 4 September 2023) <http://arxiv.org/abs/2307.03718> accessed 15 September 2023.
[5] ‘A Law for Foundation Models: The EU AI Act Can Improve Regulation for Fairer Competition – OECD.AI’ <https://oecd.ai/en/wonk/foundation-models-eu-ai-act-fairer-competition> accessed 15 September 2023.
[6] ‘Stanford CRFM’ <https://crfm.stanford.edu/report.html> accessed 15 September 2023.
[7] ‘While only a few well-resourced actors worldwide have released general purpose AI models, hundreds of millions of end-users already use these models, further scaled by potentially thousands of applications building on them across a variety of sectors, ranging from education and healthcare to media and finance.’ Pegah Maham and Sabrina Küspert, ‘Governing General Purpose AI’.
[8] Draft standards here are a very good example of the value of dataset documentation (i.e. declaring metadata) on what is used in training and fine-tuning models. In theory, this could also all be kept confidential as commercially sensitive information once a legal infrastructure is in place www.datadiversity.org/draft-standards
[9] Mitchell, Wu, Zaldivar, Barnes, Vasserman, Hutchinson, Spitzer, Raji and Gebru, (2019), ‘Model Cards for Model Reporting’, doi: 10.1145/3287560.3287596
[10] Gebru, Morgenstern, Vecchione, Vaughan, Wallach, Daum and Crawford, (2021), Datasheets for Datasets, https://m-cacm.acm.org/magazines/2021/12/256932-datasheets-for-datasets/abstract (Accessed: 27 February 2023) Hutchinson, Smart, Hanna, Denton, Greer, Kjartansson, Barnes and Mitchell, (2021), ‘Towards Accountability for Machine Learning Datasets: Practices from Software Engineering and Infrastructure’, doi: 10.1145/3442188.3445918;
[11] In the UK, the Civil Aviation Authority has a revenue of £140m and staff of over 1,000, and the Office for Nuclear Regulation around £90m with around 700 staff). An EU-level agency for AI should be funded well beyond this, given that the EU is more than six times the size of the UK.
[12] Algorithmic Accountability Act of 2022 <2022-02-03 Algorithmic Accountability Act of 2022 One-pager (senate.gov)> accessed 15 September 2023.
[13] Lingjiao Chen, Matei Zaharia and James Zou, ‘How Is ChatGPT’s Behavior Changing over Time?’ (arXiv, 1 August 2023) <http://arxiv.org/abs/2307.09009> accessed 15 September 2023.
[14] ‘AI-Generated Books on Amazon Could Give Deadly Advice – Decrypt’ <https://decrypt.co/154187/ai-generated-books-on-amazon-could-give-deadly-advice> accessed 15 September 2023.
[15] ‘Generative AI for Medical Research | The BMJ’ <www.bmj.com/content/382/bmj.p1551#> accessed 15 September 2023.
[16] Emanuel Maiberg ·, ‘Inside the AI Porn Marketplace Where Everything and Everyone Is for Sale’ (404 Media, 22 August 2023) <www.404media.co/inside-the-ai-porn-marketplace-where-everything-and-everyone-is-for-sale/> accessed 15 September 2023.
[17] Belle Lin, ‘AI Is Generating Security Risks Faster Than Companies Can Keep Up’ Wall Street Journal (10 August 2023) <www.wsj.com/articles/ai-is-generating-security-risks-faster-than-companies-can-keep-up-a2bdedd4> accessed 15 September 2023.
[18] Sarah Carter et. al., <The Convergence of Artificial Intelligence and the Life Sciences www.nti.org/analysis/articles/the-convergence-of-artificial-intelligence-and-the-life-sciences/> accessed 2 November 2023
[19] Dual Use of Artificial Intelligence-powered Drug Discovery – PubMed (nih.gov)
[20] Haydn Belfield, ‘Great British Cloud And BritGPT: The UK’s AI Industrial Strategy Must Play To Our Strengths’ (Labour for the Long Term 2023)
[21] Thinking About Risks From AI: Accidents, Misuse and Structure | Lawfare (lawfaremedia.org)
[22] Governing General Purpose AI — A Comprehensive Map of Unreliability, Misuse and Systemic Risks | Stiftung Neue Verantwortung (SNV) (stiftung-nv.de); Anthropic \ Frontier Threats Red Teaming for AI Safety
[23] www.deepmind.com/blog/an-early-warning-system-for-novel-ai-risks
[24] ‘Mission critical: Lessons from relevant sectors for AI safety’ (Ada Lovelace Institute 2023) <https://www.adalovelaceinstitute.org/policy-briefing/ai-safety/> accessed 23 November 2023
[25] ‘EU AI Standards Development and Civil Society Participation’ <www.adalovelaceinstitute.org/event/eu-ai-standards-civil-society-participation/> accessed 18 September 2023.
[26] Algorithmic Accountability Act of 2022 <2022-02-03 Algorithmic Accountability Act of 2022 One-pager (senate.gov)> accessed 15 September 2023.
[27] ‘The Problem with AI Licensing & an “FDA for Algorithms” | The Federalist Society’ <https://fedsoc.org/commentary/fedsoc-blog/the-problem-with-ai-licensing-an-fda-for-algorithms> accessed 15 September 2023.
[28] ‘Clip: Amy Kapczynski on an Old Idea Getting New Attention–an “FDA for AI”. – AI Now Institute’ <https://ainowinstitute.org/general/clip-amy-kapczynski-on-an-old-idea-getting-new-attention-an-fda-for-ai> accessed 15 September 2023.
[29] Dylan Matthews, ‘The AI Rules That US Policymakers Are Considering, Explained’ (Vox, 1 August 2023) <www.vox.com/future-perfect/23775650/ai-regulation-openai-gpt-anthropic-midjourney-stable> accessed 15 September 2023; Belenguer L, ‘AI Bias: Exploring Discriminatory Algorithmic Decision-Making Models and the Application of Possible Machine-Centric Solutions Adapted from the Pharmaceutical Industry’ (2022) 2 AI and Ethics 771 <https://doi.org/10.1007/s43681-022-00138-8>
[30] ‘Senate Hearing on Regulating Artificial Intelligence Technology | C-SPAN.Org’ <www.c-span.org/video/?529513-1/senate-hearing-regulating-artificial-intelligence-technology> accessed 15 September 2023.
[31] ‘AI Algorithms Need FDA-Style Drug Trials | WIRED’ <www.wired.com/story/ai-algorithms-need-drug-trials/> accessed 15 September 2023.
[32] ‘One of the “Godfathers of AI” Airs His Concerns’ The Economist <www.economist.com/by-invitation/2023/07/21/one-of-the-godfathers-of-ai-airs-his-concerns> accessed 15 September 2023.
[33] ‘ISVP’ <www.senate.gov/isvp/?auto_play=false&comm=judiciary&filename=judiciary072523&poster=www.judiciary.senate.gov/assets/images/video-poster.png&stt=> accessed 15 September 2023.
{kind=link}
[34] ‘Regulations.Gov’ <www.regulations.gov/docket/NTIA-2023-0005/comments> accessed 15 September 2023.
[35] Guidelines for Artificial Intelligence in Medicine: Literature Review and Content Analysis of Frameworks – PMC (nih.gov)
[36] ‘Foundation Models for Generalist Medical Artificial Intelligence | Nature’ <www.nature.com/articles/s41586-023-05881-4> accessed 15 September 2023.
[37] Anthropic admitted openly that “we do not know how to train systems to robustly behave well“. ‘Core Views on AI Safety: When, Why, What, and How’ (Anthropic) <www.anthropic.com/index/core-views-on-ai-safety> accessed 18 September 2023.
[38] NTIA AI Accountability Request for Comment <www.regulations.gov/docket/NTIA-2023-0005/comments> accessed 18 September 2023.
[39] Inioluwa Deborah Raji and others, ‘Outsider Oversight: Designing a Third Party Audit Ecosystem for AI Governance’ (arXiv, 9 June 2022) <http://arxiv.org/abs/2206.04737> accessed 18 September 2023.
[40] See Appendix for a list of interviewees
[41] Michael Moor and others, ‘Foundation Models for Generalist Medical Artificial Intelligence’ (2023) 616 Nature 259.
[42] Lewis Ho and others, ‘International Institutions for Advanced AI’ (arXiv, 11 July 2023) <http://arxiv.org/abs/2307.04699> accessed 18 September 2023.
[43] Center for Devices and Radiological Health, ‘Medical Device Single Audit Program (MDSAP)’ (FDA, 24 August 2023) <www.fda.gov/medical-devices/cdrh-international-programs/medical-device-single-audit-program-mdsap> accessed 18 September 2023.
[44] Center for Drug Evaluation and Research, ‘Conducting Clinical Trials’ (FDA, 2 August 2023) <www.fda.gov/drugs/development-approval-process-drugs/conducting-clinical-trials> accessed 18 September 2023.
[45] ‘Explainer: What Is a Foundation Model?’ <www.adalovelaceinstitute.org/resource/foundation-models-explainer/> accessed 18 September 2023.
Alternatively: ‘any model that is trained on broad data (generally using self-supervision at scale) that can be adapted (e.g.,fine-tuned) to a wide range of downstream tasks’.
Bommasani R and others, ‘On the Opportunities and Risks of Foundation Models’ (arXiv, 12 July 2022) <http://arxiv.org/abs/2108.07258>
[46] ‘Explainer: What Is a Foundation Model?’ <www.adalovelaceinstitute.org/resource/foundation-models-explainer/> accessed 18 September 2023.
[47] Ibid.
[48] AWS, ‘Fine-Tune a Model’ <https://docs.aws.amazon.com/sagemaker/latest/dg/jumpstart-fine-tune.html> accessed 3 July 2023
[49] ‘Explainer: What Is a Foundation Model?’ <www.adalovelaceinstitute.org/resource/foundation-models-explainer/> accessed 18 September 2023.
[50] ‘ISO – ISO 9001 and Related Standards — Quality Management’ (ISO, 1 September 2021) <www.iso.org/iso-9001-quality-management.html> accessed 2 November 2023.
[51] 14:00-17:00, ‘ISO 13485:2016’ (ISO, 2 June 2021) <www.iso.org/standard/59752.html> accessed 2 November 2023.
[52] OECD, ‘Risk-Based Regulation’ in OECD, OECD Regulatory Policy Outlook 2021 (OECD 2021) <www.oecd-ilibrary.org/governance/oecd-regulatory-policy-outlook-2021_9d082a11-en> accessed 18 September 2023.
[53] Center for Devices and Radiological Health, ‘International Medical Device Regulators Forum (IMDRF)’ (FDA, 15 September 2023) <www.fda.gov/medical-devices/cdrh-international-programs/international-medical-device-regulators-forum-imdrf> accessed 18 September 2023.
[54] Office of the Commissioner, ‘What We Do’ (FDA, 28 June 2021) <www.fda.gov/about-fda/what-we-do> accessed 18 September 2023.
[55] ‘FDA User Fees: Examining Changes in Medical Product Development and Economic Benefits’ (ASPE) <https://aspe.hhs.gov/reports/fda-user-fees> accessed 18 September 2023.
[56] ‘Premarket Approval (PMA)’ <www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfpma/pma.cfm?id=P160009> accessed 18 September 2023.
[57] ‘Product Classification’ <www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfPCD/classification.cfm?id=LQB> accessed 18 September 2023.
[58] Center for Devices and Radiological Health, ‘Et Control – P210018’ [2022] FDA <www.fda.gov/medical-devices/recently-approved-devices/et-control-p210018> accessed 18 September 2023.
[59] Note that only ~2% of SaMD are Class III, see Approval of artificial intelligence and machine learning-based medical devices in the USA and Europe (2015–20): a comparative analysis – The Lancet Digital Health and Drugs and Devices: Comparison of European and U.S. Approval Processes – ScienceDirect
[60] ‘Assessing the Efficacy and Safety of Medical Technologies (Part 4 of 12) (Princeton.Edu) – Google Search’ <www.google.com/search?q=Assessing+the+Efficacy+and+Safety+of+Medical+Technologies+(Part+4+of+12)+(princeton.edu)&rlz=1C1GCEA_enBE1029BE1030&oq=Assessing+the+Efficacy+and+Safety+of+Medical+Technologies+(Part+4+of+12)+(princeton.edu)&gs_lcrp=EgZjaHJvbWUyBggAEEUYOdIBBzM1N2owajSoAgCwAgA&sourceid=chrome&ie=UTF-8> accessed 18 September 2023.
[61] Ibid.
[62] For the purposes of this report, ‘effectiveness’ is used as a synonym of ‘efficacy’. In detail, effectiveness is concerned with the benefit of a technology under average conditions of use, whereas efficacy is the benefit under ideal conditions.
[63] ‘SAMD MDSW’ <www.quaregia.com/blog/samd-mdsw> accessed 18 September 2023.
[64] Office of the Commissioner, ‘The Drug Development Process’ (FDA, 20 February 2020) <www.fda.gov/patients/learn-about-drug-and-device-approvals/drug-development-process> accessed 18 September 2023.
[65] Eric Wu and others, ‘How Medical AI Devices Are Evaluated: Limitations and Recommendations from an Analysis of FDA Approvals’ (2021) 27 Nature Medicine 582.
[66] It can be debated whether this falls under the exact definition of SaMD as a stand-alone software feature, or as a software component of a medical device, but the lessons and process remain the same.
[67] SUMMARY OF SAFETYAND EFFECTIVENESS DATA (SSED) <www.accessdata.fda.gov/cdrh_docs/pdf21/P210018B.pdf> accessed 18 September 2023.
[68] A QMS is a standardised process for documenting compliance based on international standards (ISO 13485/820).
[69] Center for Devices and Radiological Health, ‘Overview of IVD Regulation’ [2023] FDA <www.fda.gov/medical-devices/ivd-regulatory-assistance/overview-ivd-regulation> accessed 18 September 2023.
[70] ‘When Science and Politics Collide: Enhancing the FDA | Science’ <www.science.org/doi/10.1126/science.aaw8093> accessed 18 September 2023.
[71] ‘Unique Device Identification System’ (Federal Register, 24 September 2013) <www.federalregister.gov/documents/2013/09/24/2013-23059/unique-device-identification-system> accessed 18 September 2023.
[72] ‘openFDA’ <https://open.fda.gov/data/faers/> accessed 10 November 2023.
[73] For example, Carpenter 2010, Hilts 2004, Hutt et al 2022
[74] ‘Factors to Consider Regarding Benefit-Risk in Medical Device Product Availability, Compliance, and Enforcement Decisions – Guidance for Industry and Food and Drug Administration Staff’.
[75] Center for Devices and Radiological Health, ‘510(k) Third Party Review Program’ (FDA, 15 August 2023) <www.fda.gov/medical-devices/premarket-submissions-selecting-and-preparing-correct-submission/510k-third-party-review-program> accessed 18 September 2023.
[76] Office of Regulatory Affairs, ‘What Should I Expect during an Inspection?’ [2020] FDA <www.fda.gov/industry/fda-basics-industry/what-should-i-expect-during-inspection> accessed 18 September 2023.
[77] ‘Device Makers Can Take COTS, but Only with Clear SOUP’ <https://web.archive.org/web/20130123140527/http://medicaldesign.com/engineering-prototyping/software/device-cots-soup-1111/> accessed 18 September 2023.
[78] ‘FDA Clears Intellia to Start US Tests of “in Vivo” Gene Editing Drug’ (BioPharma Dive) <www.biopharmadive.com/news/intellia-fda-crispr-in-vivo-gene-editing-ind/643999/> accessed 18 September 2023.
[79] ‘FDA Authority Over Tobacco’ (Campaign for Tobacco-Free Kids) <www.tobaccofreekids.org/what-we-do/us/fda> accessed 18 September 2023.
[80] FDA AT A GLANCE: REGULATED PRODUCTS AND FACILITIES, November 2020 <www.fda.gov/media/143704/download#:~:text=REGULATED%20PRODUCTS%20AND%20FACILITIES&text=FDA%2Dregulated%20products%20account%20for,dollar%20spent%20by%20U.S.%20consumers.&text=FDA%20regulates%20about%2078%20percent,poultry%2C%20and%20some%20egg%20products.> accessed 18 September 2023.
[81] ‘Getting Smarter: FDA Publishes Draft Guidance on Predetermined Change Control Plans for Artificial Intelligence/Machine Learning (AI/ML) Devices’ (5 February 2023) <www.ropesgray.com/en/newsroom/alerts/2023/05/getting-smarter-fda-publishes-draft-guidance-on-predetermined-change-control-plans-for-ai-ml-devices> accessed 18 September 2023.
[82] Center for Veterinary Medicine, ‘Q&A on FDA Regulation of Intentional Genomic Alterations in Animals’ [2023] FDA <www.fda.gov/animal-veterinary/intentional-genomic-alterations-igas-animals/qa-fda-regulation-intentional-genomic-alterations-animals> accessed 18 September 2023.
[83] Andrew Kolodny, ‘How FDA Failures Contributed to the Opioid Crisis’ (2020) 22 AMA Journal of Ethics 743.
[84] Commissioner O of the, ‘Milestones in U.S. Food and Drug Law’ [2023] FDA <https://www.fda.gov/about-fda/fda-history/milestones-us-food-and-drug-law> accessed 3 December 2023
[85] Reputation and Power (2010) <https://press.princeton.edu/books/paperback/9780691141800/reputation-and-power> accessed 3 December 2023
[86] ‘Hutt, Merrill, Grossman, Cortez, Lietzan, and Zettler’s Food and Drug Law, 5th – 9781636596952 – West Academic’ <https://faculty.westacademic.com/Book/Detail?id=341299> accessed 3 December 2023
[87]For example Carpenter 2010, Hilts 2004, Hutt et al 2022
[88] ‘Hutt, Merrill, Grossman, Cortez, Lietzan, and Zettler’s Food and Drug Law, 5th – 9781636596952 – West Academic’ <https://faculty.westacademic.com/Book/Detail?id=341299> accessed 18 September 2023.
[89] Eric Wu and others, ‘How Medical AI Devices Are Evaluated: Limitations and Recommendations from an Analysis of FDA Approvals’ (2021) 27 Nature Medicine 582.
[90] Other public health regulators, for example NICE (UK) cover accessibility risk to a larger degree than the FDA, similarly on structural discrimination risks with NICE “Standing together” work on data curation and declarations of datasets used in developing SaMD. The FDA over time developed similar programs.
[91] Ziad Obermeyer and others, ‘Dissecting Racial Bias in an Algorithm Used to Manage the Health of Populations’ (2019) 366 Science 447.
[92] ’FDA-cleared artificial intelligence and machine learning-based medical devices and their 510(k) predicate networks’<www.thelancet.com/journals/landig/article/PIIS2589-7500(23)00126-7/fulltext#sec1> accessed 18 September 2023.
[93] ‘How the FDA’s Food Division Fails to Regulate Health and Safety Hazards’ <https://politico.com/interactives/2022/fda-fails-regulate-food-health-safety-hazards> accessed 18 September 2023.
[94] Christopher J Morten and Amy Kapczynski, ‘The Big Data Regulator, Rebooted: Why and How the FDA Can and Should Disclose Confidential Data on Prescription Drugs and Vaccines’ (2021) 109 California Law Review 493.
[95] ‘Examination of Clinical Trial Costs and Barriers for Drug Development’ (ASPE) <https://aspe.hhs.gov/reports/examination-clinical-trial-costs-barriers-drug-development-0> accessed 18 September 2023.
[96] Office of the Commissioner, ‘Advisory Committees’ (FDA, 3 May 2021) <www.fda.gov/advisory-committees> accessed 18 September 2023.
[97] For example. Carpenter 2010, Hilts 2004, Hutt et al 2022
[98] ‘FDA’s Science Infrastructure Failing | Infectious Diseases | JAMA | JAMA Network’ <https://jamanetwork.com/journals/jama/article-abstract/1149359> accessed 18 September 2023.
[99] Bridget M Kuehn, ‘FDA’s Science Infrastructure Failing’ (2008) 299 JAMA 157.
[100] ‘What to Expect at FDA’s Vaccine Advisory Committee Meeting’ (The Equation, 19 October 2020) <https://blog.ucsusa.org/genna-reed/vrbpac-meeting-what-to-expect/> accessed 18 September 2023.
[101] Office of the Commissioner, ‘What Is a Conflict of Interest?’ [2022] FDA <www.fda.gov/about-fda/fda-basics/what-conflict-interest> accessed 18 September 2023.
[102] The Firm and the FDA: McKinsey & Company’s Conflicts of Interest at the Heart of the Opioid Epidemic <https://fingfx.thomsonreuters.com/gfx/legaldocs/akpezyejavr/2022-04-13.McKinsey%20Opioid%20Conflicts%20Majority%20Staff%20Report%20FINAL.pdf> accessed 18 September 2023.
[103] Causholli M, Chambers DJ and Payne JL, ‘Future Nonaudit Service Fees and Audit Quality’ (2014) ,<onlinelibrary.wiley.com/doi/abs/10.1111/1911-3846.12042> accessed 21 September 2023; Jamal K and Sunder S, ‘Is Mandated Independence Necessary for Audit Quality?’ (2011) 36 Accounting, Organizations and Society 284 <Is mandated independence necessary for audit quality? – ScienceDirect> accessed 21 September 2023
[104] Reputation and Power (2010) <https://press.princeton.edu/books/paperback/9780691141800/reputation-and-power> accessed 18 September 2023.
[105] ‘Hutt, Merrill, Grossman, Cortez, Lietzan, and Zettler’s Food and Drug Law, 5th – 9781636596952 – West Academic’ <https://faculty.westacademic.com/Book/Detail?id=341299> accessed 18 September 2023.
[106] Ana Santos Rutschman, ‘How Theranos’ Faulty Blood Tests Got to Market – and What That Shows about Gaps in FDA Regulation’ (The Conversation, 5 October 2021) <http://theconversation.com/how-theranos-faulty-blood-tests-got-to-market-and-what-that-shows-about-gaps-in-fda-regulation-168050> accessed 18 September 2023.
[107] Center for Devices and Radiological Health, ‘Classify Your Medical Device’ (FDA, 14 August 2023) <www.fda.gov/medical-devices/overview-device-regulation/classify-your-medical-device> accessed 18 September 2023.
[108] Anderljung and others, ‘Frontier AI Regulation: Managing Emerging Risks to Public Safety’ (arXiv, 4 September 2023) <http://arxiv.org/abs/2307.03718> accessed 15 September 2023.
[109] ‘A Law for Foundation Models: The EU AI Act Can Improve Regulation for Fairer Competition – OECD.AI’ <https://oecd.ai/en/wonk/foundation-models-eu-ai-act-fairer-competition> accessed 18 September 2023.
[110] ‘Stanford CRFM’ <https://crfm.stanford.edu/report.html> accessed 18 September 2023.
[111] Pegah Maham and Sabrina Küspert, ‘Governing General Purpose AI’.
[112] ‘Frontier AI Regulation: Managing Emerging Risks to Public Safety’ <https://openai.com/research/frontier-ai-regulation> accessed 18 September 2023.
[113] ‘Auditing Algorithms: The Existing Landscape, Role of Regulators and Future Outlook’ (GOV.UK) <www.gov.uk/government/publications/findings-from-the-drcf-algorithmic-processing-workstream-spring-2022/auditing-algorithms-the-existing-landscape-role-of-regulators-and-future-outlook> accessed 18 September 2023.
[114] ‘Introducing Superalignment’ <https://openai.com/blog/introducing-superalignment> accessed 18 September 2023.
[115] ‘Why AI Safety?’ (Machine Intelligence Research Institute) <https://intelligence.org/why-ai-safety/> accessed 18 September 2023.
[116] ‘DAIR (Distributed AI Research Institute)’ (DAIR Institute) <https://dair-institute.org/> accessed 18 September 2023.
[117] Anthropic < https://www.anthropic.com/index/frontier-threats-red-teaming-for-ai-safety#:~:text=If%20unmitigated%2C%20we%20worry%20that,implementation%20of%20mitigations%20for%20them> accessed 29 November 2023
[118] ‘Explainer: What Is a Foundation Model?’ <www.adalovelaceinstitute.org/resource/foundation-models-explainer/> accessed 18 September 2023.
[119] Center for Devices and Radiological Health, ‘Software as a Medical Device (SaMD)’ (FDA, 9 September 2020) <www.fda.gov/medical-devices/digital-health-center-excellence/software-medical-device-samd> accessed 10 November 2023.
[120] Pegah Maham and Sabrina Küspert, ‘Governing General Purpose AI’.
[121] ‘The Human Decisions That Shape Generative AI’ (Mozilla Foundation, 2 August 2023) <https://foundation.mozilla.org/en/blog/the-human-decisions-that-shape-generative-ai-who-is-accountable-for-what/> accessed 18 September 2023.
[122] ‘Frontier Model Security’ (Anthropic) <www.anthropic.com/index/frontier-model-security> accessed 18 September 2023.
[123] Is ChatGPT a cybersecurity threat? | TechCrunch
[124] ChatGPT Security Risks: What Are They and How To Protect Companies (itprotoday.com)
[125] 2307.03718.pdf (arxiv.org)
[126] 2307.03718.pdf (arxiv.org)
[127] 2307.03718.pdf (arxiv.org)
[128] 2307.03718.pdf (arxiv.org)
[129] ‘AI Assurance?’ <www.adalovelaceinstitute.org/report/risks-ai-systems/> accessed 21 September 2023.
[130] Preparing for Extreme Risks: Building a Resilient Society (parliament.uk) ‘Preparing for Extreme Risks: Building a Resilient Society’
[131] Nguyen T, ‘Insurability of Catastrophe Risks and Government Participation in Insurance Solutions’ (2013) <www.semanticscholar.org/paper/Insurability-of-Catastrophe-Risks-and-Government-in-Nguyen/dcecefd3f24a099b958e8ac1127a4bdc803b28fb> accessed 21 September 2023
[132] Banias MJ, ‘Inside CounterCloud: A Fully Autonomous AI Disinformation System’ (The Debrief, 16 August 2023) <https://thedebrief.org/countercloud-ai-disinformation/> accessed 21 September 2023
[133] Raji ID and others, ‘Outsider Oversight: Designing a Third Party Audit Ecosystem for AI Governance’ (arXiv, 9 June 2022) <http://arxiv.org/abs/2206.04737> accessed 21 September 2023
[134] McAllister LK, ‘Third-Party Programs to Assess Regulatory Compliance’ (2012) <www.acus.gov/sites/default/files/documents/Third-Party-Programs-Report_Final.pdf> accessed 21 September 2023
[135] Science in Regulation, A Study of Agency Decisionmaking Approaches, Appendices 2012 <www.acus.gov/sites/default/files/documents/Science%20in%20Regulation_Final%20Appendix_2_18_13_0.pdf> accessed 21 September 2023
[136] GPT-4-system-card (openai.com) (2023) <https://cdn.openai.com/papers/gpt-4-system-card.pdf> accessed 21 September 2023
[137] Intensive own evidence production of regulators, for example like the IAEA, is only suitable for non-complex industries
[138] The order does not indicate the importance of each dimension. The importance for risk reduction depends significantly on the specific implementation of the dimensions and the context.
[139] While other oversight regimes such as practised in cybersecurity, aviation or similar are an inspiration for foundation models too, FDA-style oversight is among the few that score towards the right on most dimensions identified in the regulatory oversight and audit literature and depicted above.
[140] Open AI Bug Bounty Program (2022) <Announcing OpenAI’s Bug Bounty Program> accessed 21 September 2023
[141] ‘MAUDE – Manufacturer and User Facility Device Experience’ <www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfmaude/search.cfm> accessed 21 September 2023
[142] ‘Auditor Independence and Audit Quality: A Literature Review – Nopmanee Tepalagul, Ling Lin, 2015’ <https://journals.sagepub.com/doi/abs/10.1177/0148558×14544505?casa_token=6R7ABlbi2I0AAAAA:K1pMF6sw6QrmvEhczXbW0BwjE8xXD0r3GKfOHpZczbeIvdMckGn00I6zkluRqd06WmBJXJ616xz_KXk> accessed 21 September 2023
[143] ‘Customer-Driven Misconduct: How Competition Corrupts Business Practices – Article – Faculty & Research – Harvard Business School’ <www.hbs.edu/faculty/Pages/item.aspx?num=43347> accessed 21 September 2023
[144] Donald R. Deis Jr and Giroux GA, ‘Determinants of Audit Quality in the Public Sector’ (1992) 67 The Accounting Review 462 <www.jstor.org/stable/247972?casa_token=luGLXHQ3nAoAAAAA:clOnnu3baxAfZYMCx7kJloL08GI0RPboKMovVPQz7Z6bi9w4grsJEqz1tNIKJD88yFXbpc8iqLDoeZY9U5jnECBH99hKFWKk3-WxI9e__HBwlQ_bOBhSWQ> accessed 21 September 2023
[145] Engstrom DF and Ho DE, ‘Algorithmic Accountability in the Administrative State’ (9 March 2020) <https://papers.ssrn.com/abstract=3551544> accessed 21 September 2023
[146] Causholli M, Chambers DJ and Payne JL, ‘Future Nonaudit Service Fees and Audit Quality’ (2014) ,<onlinelibrary.wiley.com/doi/abs/10.1111/1911-3846.12042> accessed 21 September 2023
[147] Jamal K and Sunder S, ‘Is Mandated Independence Necessary for Audit Quality?’ (2011) 36 Accounting, Organizations and Society 284 <Is mandated independence necessary for audit quality? – ScienceDirect> accessed 21 September 2023
[148] Widder DG, West S and Whittaker M, ‘Open (For Business): Big Tech, Concentrated Power, and the Political Economy of Open AI’ (17 August 2023) <https://papers.ssrn.com/abstract=4543807> accessed 21 September 2023
[149] Lamoreaux PT, ‘Does PCAOB Inspection Access Improve Audit Quality? An Examination of Foreign Firms Listed in the United States’ (2016) 61 Journal of Accounting and Economics 313
<Does PCAOB inspection access improve audit quality? An examination of foreign firms listed in the United States – ScienceDirect> accessed 21 September 2023
[150] ‘Introduction to NIST FRVT’ (Paravision) <www.paravision.ai/news/introduction-to-nist-frvt/> accessed 21 September 2023
[151] ‘Confluence Mobile – UN Statistics Wiki’ <https://unstats.un.org/wiki/plugins/servlet/mobile?contentId=152797274#content/view/152797274> accessed 21 September 2023
[152] ‘Large Language Models and Software as a Medical Device – MedRegs’ <https://medregs.blog.gov.uk/2023/03/03/large-language-models-and-software-as-a-medical-device/> accessed 21 September 2023
[153] Ada Lovelace Institute, AI assurance? Assessing and mitigating risks across the AI lifecycle (2023) < https://www.adalovelaceinstitute.org/report/risks-ai-systems/>
[154] ‘Inclusive AI Governance – Ada Lovelace Institute’ (2023) < www.adalovelaceinstitute.org/wp-content/uploads/2023/03/Ada-Lovelace-Institute-Inclusive-AI-governance-Discussion-paper-March-2023.pdf> accessed 21 September 2023
[155] ‘AI Assurance?’ <www.adalovelaceinstitute.org/report/risks-ai-systems/> accessed 21 September 2023
[156] ‘Comment of the AI Policy and Governance Working Group on the NTIA AI Accountability Policy’ (2023) <www.ias.edu/sites/default/files/AI%20Policy%20and%20Governance%20Working%20Group%20NTIA%20Comment.pdf> accessed 21 September 2023
[157] Weale S and correspondent SWE, ‘Lecturers Urged to Review Assessments in UK amid Concerns over New AI Tool’ The Guardian (13 January 2023) <https://www.theguardian.com/technology/2023/jan/13/end-of-the-essay-uk-lecturers-assessments-chatgpt-concerns-ai> accessed 23 November 2023
[158] ‘Proposing a Foundation Model Information-Sharing Regime for the UK | GovAI Blog’ <www.governance.ai/post/proposing-a-foundation-model-information-sharing-regime-for-the-uk> accessed 21 September 2023
[159] ‘Proposing a Foundation Model Information-Sharing Regime for the UK | GovAI Blog’ <www.governance.ai/post/proposing-a-foundation-model-information-sharing-regime-for-the-uk> accessed 21 September 2023
[160] ‘Regulating AI in the UK’ <www.adalovelaceinstitute.org/report/regulating-ai-in-the-uk/> accessed 21 September 2023
[161] ‘Unique Device Identification System’ (Federal Register, 24 September 2013) <www.federalregister.gov/documents/2013/09/24/2013-23059/unique-device-identification-system> accessed 21 September 2023
[162] Anthropic AB is CL at and others, ‘How We Can Regulate AI—Asterisk’ <https://asteriskmag.com/issues/03/how-we-can-regulate-ai> accessed 21 September 2023
[163] ‘Opinion | Here’s a Simple Way to Regulate Powerful AI Models’ Washington Post (16 August 2023) <www.washingtonpost.com/opinions/2023/08/16/ai-danger-regulation-united-states/> accessed 21 September 2023
[164] Vidal DE and others, ‘Navigating US Regulation of Artificial Intelligence in Medicine—A Primer for Physicians’ (2023) 1 Mayo Clinic Proceedings: Digital Health 31
[165] ‘The Human Decisions That Shape Generative AI’ (Mozilla Foundation, 2 August 2023) <https://foundation.mozilla.org/en/blog/the-human-decisions-that-shape-generative-ai-who-is-accountable-for-what/> accessed 21 September 2023
[166] Birhane A, Prabhu VU and Kahembwe E, ‘Multimodal Datasets: Misogyny, Pornography, and Malignant Stereotypes’ (arXiv, 5 October 2021) <http://arxiv.org/abs/2110.01963> accessed 21 September 2023
[167] Schaul K, Chen SY and Tiku N, ‘Inside the Secret List of Websites That Make AI like ChatGPT Sound Smart’ (Washington Post) <www.washingtonpost.com/technology/interactive/2023/ai-chatbot-learning/> accessed 21 September 2023
[168] ‘When AI Is Trained on AI-Generated Data, Strange Things Start to Happen’ (Futurism) <https://futurism.com/ai-trained-ai-generated-data-interview> accessed 21 September 2023
[169] Draft standards here are a very good example of the value of dataset documentation (that is, declaring metadata) on what is used in training and fine-tuning models. In theory, this could also all be kept confidential as commercially sensitive information once a legal infrastructure is in place www.datadiversity.org/draft-standards
[170] Mitchell, Wu, Zaldivar, Barnes, Vasserman, Hutchinson, Spitzer, Raji and Gebru, (2019), ‘Model Cards for Model Reporting’, doi: 10.1145/3287560.3287596
[171] Gebru, Morgenstern, Vecchione, Vaughan, Wallach, Daum and Crawford, (2021), Datasheets for Datasets, <https://m-cacm.acm.org/magazines/2021/12/256932-datasheets-for-datasets/abstract >(Accessed: 27 February 2023); Hutchinson, Smart, Hanna, Denton, Greer, Kjartansson, Barnes and Mitchell, (2021), ‘Towards Accountability for Machine Learning Datasets: Practices from Software Engineering and Infrastructure’, doi: 10.1145/3442188.3445918;
[172] Shevlane T and others, ‘Model Evaluation for Extreme Risks’ (arXiv, 24 May 2023) <http://arxiv.org/abs/2305.15324> accessed 21 September 2023
[173] A pretrained AI model is a deep learning model that is already trained on large datasets to accomplish a specific task, meaning there are design choices which affect its output and performance (according to one leading lab ‘language models already learn a lot about human values during pretraining’ and this is where ‘implicit biases’ arise.)
[174] ‘running against a suite of benchmark objectionable behaviors… we find that the prompts achieve up to 84% success rates at attacking GPT-3.5 and GPT-4, and 66% for PaLM-2; success rates for Claude are substantially lower (2.1%), but notably the attacks still can induce behavior that is otherwise never generated.’ Zou A and others, ‘Universal and Transferable Adversarial Attacks on Aligned Language Models’ (arXiv, 27 July 2023) <http://arxiv.org/abs/2307.15043> accessed 21 September 2023
[175] Shevlane T and others, ‘Model Evaluation for Extreme Risks’ (arXiv, 24 May 2023) <http://arxiv.org/abs/2305.15324> accessed 21 September 2023; Nelson et al ; Kolt N, ‘Algorithmic Black Swans’ (25 February 2023) <https://papers.ssrn.com/abstract=4370566> accessed 21 September 2023
[176] Mökander J and others, ‘Auditing Large Language Models: A Three-Layered Approach’ [2023] AI and Ethics <http://arxiv.org/abs/2302.08500> accessed 21 September 2023; Wan A and others, ‘Poisoning Language Models During Instruction Tuning’ (arXiv, 1 May 2023) <http://arxiv.org/abs/2305.00944> accessed 21 September 2023; ‘Analyzing the European Union AI Act: What Works, What Needs Improvement’ (Stanford HAI) <https://hai.stanford.edu/news/analyzing-european-union-ai-act-what-works-what-needs-improvement> accessed 21 September 2023; ‘EU AI Standards Development and Civil Society Participation’ <www.adalovelaceinstitute.org/event/eu-ai-standards-civil-society-participation/> accessed 21 September 2023
[177] ’Outsider Oversight: Designing a Third Party Audit Ecosystem for AI Governance’ <https://dl.acm.org/doi/pdf/10.1145/3514094.3534181> accessed 21 September 2023
[178] Gupta A, ‘Emerging AI Governance Is an Opportunity for Business Leaders to Accelerate Innovation and Profitability’ (Tech Policy Press, 31 May 2023) <https://techpolicy.press/emerging-ai-governance-is-an-opportunity-for-business-leaders-to-accelerate-innovation-and-profitability/> accessed 21 September 2023
[179] Key Enforcement Issues of the AI Act Should Lead EU Trilogue Debate’ (Brookings) <www.brookings.edu/articles/key-enforcement-issues-of-the-ai-act-should-lead-eu-trilogue-debate/> accessed 21 September 2023
[180] ‘Structured Access’ – Toby Shevlane (2022)< https://arxiv.org/ftp/arxiv/papers/2201/2201.05159.pdf> accessed 21 September 2023
[181] ‘Systematic probing of an AI model or system by either expert or non-expert human evaluators to reveal undesired outputs or behaviors’.
[182] House TW, ‘FACT SHEET: Biden-Harris Administration Secures Voluntary Commitments from Leading Artificial Intelligence Companies to Manage the Risks Posed by AI’ (The White House, 21 July 2023) <www.whitehouse.gov/briefing-room/statements-releases/2023/07/21/fact-sheet-biden-harris-administration-secures-voluntary-commitments-from-leading-artificial-intelligence-companies-to-manage-the-risks-posed-by-ai/> accessed 21 September 2023
[183] ‘Keeping an Eye on AI’ <www.adalovelaceinstitute.org/report/keeping-an-eye-on-ai/> accessed 21 September 2023
[184].Janjeva A and others, ‘Strengthening Resilience to AI Risk’ (2023 <)https://cetas.turing.ac.uk/sites/default/files/2023-08/cetas-cltr_ai_risk_briefing_paper.pdf> accessed 21 September 2023
[185] Shrishak K, ‘How to Deal with an AI Near-Miss: Look to the Skies’ (2023) 79 Bulletin of the Atomic Scientists 166
[186] ‘Guidance for Manufacturers on Reporting Adverse Incidents Involving Software as a Medical Device under the Vigilance System’ (GOV.UK) <www.gov.uk/government/publications/reporting-adverse-incidents-involving-software-as-a-medical-device-under-the-vigilance-system/guidance-for-manufacturers-on-reporting-adverse-incidents-involving-software-as-a-medical-device-under-the-vigilance-system> accessed 21 September 2023
[187] www.adalovelaceinstitute.org/blog/ai-regulation-learn-from-history/ Guidance always has its roots in legislation, but can be iterated more rapidly and flexibly whereas legislation requires several legal and political steps at minimum. Explainer here: www.oneeducation.org.uk/difference-between-laws-regulations-acts-guidance-policies/.
[188] www.tandfonline.com/doi/pdf/10.1080/01972243.2022.2124565?needAccess=true
[189] https://cip.org/alignmentassemblies
[190] https://arxiv.org/abs/2306.09871 ; https://openai.com/blog/democratic-inputs-to-ai
[191] Ada Lovelace Institute, Participatory data stewardship: A framework for involving people in the use of data’ (2021) < https://www.adalovelaceinstitute.org/report/participatory-data-stewardship/>
[192] Shevlane T and others, ‘Model Evaluation for Extreme Risks’ (arXiv, 24 May 2023) <http://arxiv.org/abs/2305.15324> accessed 21 September 2023
[193] ‘Examining the Black Box’ <www.adalovelaceinstitute.org/report/examining-the-black-box-tools-for-assessing-algorithmic-systems/> accessed 21 September 2023
[194] Nelson and et al., ‘AI Policy and Governance Working Group NTIA Comment.Pdf’ <www.ias.edu/sites/default/files/AI%20Policy%20and%20Governance%20Working%20Group%20NTIA%20Comment.pdf> accessed 21 September 2023
[195] Bill Chappell, ‘“It Was Installed For This Purpose,” VW’s U.S. CEO Tells Congress About Defeat Device’ NPR (8 October 2015) <www.npr.org/sections/thetwo-way/2015/10/08/446861855/volkswagen-u-s-ceo-faces-questions-on-capitol-hill> accessed 30 August 2023
[196] MedWatch is the FDA’s adverse event reporting program, while Medical Product Safety Network (MedSun) monitors the safety and effectiveness of medical devices. Commissioner O of the, ‘Step 5: FDA Post-Market Device Safety Monitoring’ [2018] FDA <www.fda.gov/patients/device-development-process/step-5-fda-post-market-device-safety-monitoring> accessed 21 September 2023
[197] AINOW, ‘Zero-Trust-AI-Governance.Pdf’ (August 2023) <https://ainowinstitute.org/wp-content/uploads/2023/08/Zero-Trust-AI-Governance.pdf> accessed 21 September 2023
[198] ‘The Value Chain of General-Purpose AI’ <www.adalovelaceinstitute.org/blog/value-chain-general-purpose-ai/> accessed 21 September 2023
[199] www.whitehouse.gov/briefing-room/statements-releases/2023/07/21/fact-sheet-biden-harris-administration-secures-voluntary-commitments-from-leading-artificial-intelligence-companies-to-manage-the-risks-posed-by-ai/
[200] Knott A and Pedreschi D, ‘State-of-the-Art Foundation AI Models Should Be Accompanied by Detection Mechanisms as a Condition of Public Release’ <https://gpai.ai/projects/responsible-ai/social-media-governance/Social%20Media%20Governance%20Project%20-%20July%202023.pdf> accessed 21 September 2023
[201] www.tspa.org/curriculum/ts-fundamentals/transparency-report/
[202] Bommasani R and others, ‘Do Foundation Model Providers Comply with the Draft EU AI Act?’ <https://crfm.stanford.edu/2023/06/15/eu-ai-act.html> accessed 21 September 2023
[203] ‘Keeping an Eye on AI’ <www.adalovelaceinstitute.org/report/keeping-an-eye-on-ai/> accessed 21 September 2023
[204] ‘Regulating AI in the UK’ <www.adalovelaceinstitute.org/report/regulating-ai-in-the-uk/> accessed 21 September 2023
[205] Zinchenko V and others, ‘Changes in Software as a Medical Device Based on Artificial Intelligence Technologies’ (2022) 17 International Journal of Computer Assisted Radiology and Surgery 1969
[206] Shrishak K, ‘How to Deal with an AI Near-Miss: Look to the Skies’ (2023) 79 Bulletin of the Atomic Scientists 166
[207] AINOW, ‘Zero-Trust-AI-Governance.Pdf’ (August 2023) <https://ainowinstitute.org/wp-content/uploads/2023/08/Zero-Trust-AI-Governance.pdf> accessed 21 September 2023
[208] ‘How Boeing 737 MAX’s Flawed Flight Control System Led to 2 Crashes That Killed 346 – ABC News’ <https://abcnews.go.com/US/boeing-737-maxs-flawed-flight-control-system-led/story?id=74321424> accessed 21 September 2023
[209] A new national system to more quickly spot possible safety issues, using existing electronic health databases to keep an eye on the safety of approved medical products in real time. This tool will add to, but not replace, FDA’s existing post-market safety assessment tools. Commissioner of the, ‘Step 5: FDA Post-Market Device Safety Monitoring’ [2018] FDA <www.fda.gov/patients/device-development-process/step-5-fda-post-market-device-safety-monitoring> accessed 21 September 2023
[210] In the UK, the Civil Aviation Authority has a revenue of £140m and staff of over 1,000, and the Office for Nuclear Regulation around £90m with around 700 staff. An EU-level agency for AI should be funded well beyond this, given that the EU is more than six times the size of the UK.
[211] Affairs O of R, ‘Recalls, Market Withdrawals, & Safety Alerts’ (FDA, 11 February 2022) <www.fda.gov/safety/recalls-market-withdrawals-safety-alerts> accessed 21 September 2023
[212] Team NA, ‘NIST AIRC – Govern’ <https://airc.nist.gov/AI_RMF_Knowledge_Base/Playbook/Govern> accessed 21 September 2023
[213]‘Committing to Effective Whistleblower Protection | En | OECD’ <www.oecd.org/corruption-integrity/reports/committing-to-effective-whistleblower-protection-9789264252639-en.html> accessed 21 September 2023
[214] Anderljung M and others, ‘Frontier AI Regulation: Managing Emerging Risks to Public Safety’ (arXiv, 4 September 2023) <http://arxiv.org/abs/2307.03718> accessed 21 September 2023
[215] Guidance always has its roots in legislation but can be iterated more rapidly and flexibly, whereas legislation requires several legal and political steps at minimum. ‘AI Regulation and the Imperative to Learn from History’ <www.adalovelaceinstitute.org/blog/ai-regulation-learn-from-history/> accessed 21 September 2023
Explainer here: www.oneeducation.org.uk/difference-between-laws-regulations-acts-guidance-policies/.
[216] Raji ID and others, ‘Outsider Oversight: Designing a Third Party Audit Ecosystem for AI Governance’ (arXiv, 9 June 2022) <http://arxiv.org/abs/2206.04737> accessed 21 September 2023
[217] Draft standards here are a very good example of the value of dataset documentation (i.e. declaring metadata) on what is used in training and fine-tuning models. In theory, this could also all be kept confidential as commercially sensitive information once a legal infrastructure is in place. www.datadiversity.org/draft-standards
[218] Mitchell, Wu, Zaldivar, Barnes, Vasserman, Hutchinson, Spitzer, Raji and Gebru, (2019), ‘Model Cards for Model Reporting’, doi: 10.1145/3287560.3287596
[219] Gebru, Morgenstern, Vecchione, Vaughan, Wallach, Daum and Crawford, (2021), Datasheets for Datasets, <https://m-cacm.acm.org/magazines/2021/12/256932-datasheets-for-datasets/abstract> (Accessed: 27 February 2023) Hutchinson, Smart, Hanna, Denton, Greer, Kjartansson, Barnes and Mitchell, (2021), ‘Towards Accountability for Machine Learning Datasets: Practices from Software Engineering and Infrastructure’, doi: 10.1145/3442188.3445918;
[220] Shevlane T and others, ‘Model Evaluation for Extreme Risks’ (arXiv, 24 May 2023) <http://arxiv.org/abs/2305.15324> accessed 21 September 2023
[222] Knott A and Pedreschi D, ‘State-of-the-Art Foundation AI Models Should Be Accompanied by Detection Mechanisms as a Condition of Public Release’ <https://gpai.ai/projects/responsible-ai/social-media-governance/Social%20Media%20Governance%20Project%20-%20July%202023.pdf> accessed 21 September 2023
[223] Shevlane T and others, ‘Model Evaluation for Extreme Risks’ (arXiv, 24 May 2023) <http://arxiv.org/abs/2305.15324> accessed 21 September 2023
[224] ‘Examining the Black Box’ <www.adalovelaceinstitute.org/report/examining-the-black-box-tools-for-assessing-algorithmic-systems/> accessed 21 September 2023
[225] AINOW, ‘Zero-Trust-AI-Governance.Pdf’ (August 2023) <https://ainowinstitute.org/wp-content/uploads/2023/08/Zero-Trust-AI-Governance.pdf> accessed 21 September 2023
[226] ‘Regulating AI in the UK’ <www.adalovelaceinstitute.org/report/regulating-ai-in-the-uk/> accessed 21 September 2023
[227] Shrishak K, ‘How to Deal with an AI Near-Miss: Look to the Skies’ (2023) 79 Bulletin of the Atomic Scientists 166
[228] Team NA, ‘NIST AIRC – Govern 1.7’ <https://airc.nist.gov/AI_RMF_Knowledge_Base/Playbook/Govern> accessed 21 September 2023
[229] In the UK, the Civil Aviation Authority has a revenue of £140m and staff of over 1,000, and the Office for Nuclear Regulation around £90m with around 700 staff). An EU-level agency for AI should be funded well beyond this, given that the EU is more than six times the size of the UK.
[230] In 2023 ~50% of the FDA’s ~$8bn budget was covered through mandatory fees by companies overseen by the FDA. See: < https://www.fda.gov/media/165045/download > accessed 24/11/2023
[231] 80% of the EMA’s funding comes from fees and charges levied on companies. See: EMA, “Funding,” European Medicines Agency, Sep. 17, 2018. <www.ema.europa.eu/en/about-us/how-we-work/governance-documents/funding> accessed Aug. 10, 2023
[232] ‘Governing General Purpose AI — A Comprehensive Map of Unreliability, Misuse and Systemic Risks’ (20 July 2023) <www.stiftung-nv.de/de/publikation/governing-general-purpose-ai-comprehensive-map-unreliability-misuse-and-systemic-risks> accessed 21 September 2023
[233] Nathalie Smuha: Beyond the Individual: Governing AI’s Societal Harm < https://papers.ssrn.com/sol3/papers.cfm?abstract_id=3941956 > accessed Nov. 24, 2023
[234] EMA, “Success rate for marketing authorisation applications from SMEs doubles between 2016 and 2020,” European Medicines Agency, Jun. 25, 2021 <www.ema.europa.eu/en/news/success-rate-marketing-authorisation-applications-smes-doubles-between-2016-2020> accessed Aug. 10, 2023
[235] ‘AI and Digital Regulations Service for Health and Social Care – AI Regulation Service – NHS’ <www.digitalregulations.innovation.nhs.uk/> accessed 21 September 2023
[236] EMA, “Advanced therapy medicinal products: Overview,” European Medicines Agency, Sep. 17, 2018. <www.ema.europa.eu/en/human-regulatory/overview/advanced-therapy-medicinal-products-overview> accessed Aug. 10, 2023
[237] ‘Key Enforcement Issues of the AI Act Should Lead EU Trilogue Debate’ (Brookings) <www.brookings.edu/articles/key-enforcement-issues-of-the-ai-act-should-lead-eu-trilogue-debate/> accessed 21 September 2023
[238] Infocomm Media Development Authority, Aicadium, and AI Verify Foundation, ‘Generative AI: Implications for Trust and Governance’ 2023 <https://aiverifyfoundation.sg/downloads/Discussion_Paper.pdf> accessed 21 September 2023
[239] EMA, “Transparency,” European Medicines Agency, Sep. 17, 2018 <www.ema.europa.eu/en/about-us/how-we-work/transparency> (accessed Aug. 10, 2023).
[240] ‘Governing General Purpose AI — A Comprehensive Map of Unreliability, Misuse and Systemic Risks’ (20 July 2023) <www.stiftung-nv.de/de/publikation/governing-general-purpose-ai-comprehensive-map-unreliability-misuse-and-systemic-risks> accessed 21 September 2023
[241] Ho L and others, ‘International Institutions for Advanced AI’ (arXiv, 11 July 2023) <http://arxiv.org/abs/2307.04699> accessed 21 September 2023
[242] ‘Three Regulatory Agencies: A Comparison’ <www.hmpgloballearningnetwork.com/site/frmc/articles/three-regulatory-agencies-comparison> accessed 21 September 2023
[243] ‘COVID-19 Disruptions of International Clinical Trials: Comparing Guidances Issued by FDA, EMA, MHRA and PMDA’ (4 February 2020) <www.ropesgray.com/en/newsroom/alerts/2020/04/national-authority-guidance-on-clinical-trials-during-the-covid-19-pandemic> accessed 21 September 2023
[244] Van Norman GA, ‘Drugs and Devices: Comparison of European and U.S. Approval Processes’ (2016) 1 JACC: Basic to Translational Science 399
[245] Cummings ML and Britton D, ‘Chapter 6 – Regulating Safety-Critical Autonomous Systems: Past, Present, and Future Perspectives’ in Richard Pak, Ewart J de Visser and Ericka Rovira (eds), Living with Robots (Academic Press 2020) <www.sciencedirect.com/science/article/pii/B9780128153673000062> accessed 21 September 2023
Image credit: Lyndon Stratford
Related content

Foundation models in the public sector
AI foundation models are integrated into commonly used applications and are used informally in the public sector